Mol Cell| The Wensheng Wei team has achieved the functional decoding of lysine residues in the human proteome
Amino acid (AA) residues are protein building blocks that underpin protein structure and activity. Genetic substitutions of AAs are widely associated with physiological changes or diseases and take up a large proportion among all the known pathogenic mutations. Although currently, base editors1,2 can achieve base substitutions in the genome, thereby altering codons and generating mutations in endogenous amino acids, systematic functional analysis of specific amino acid residues within the entire proteome still faces challenges.
On November 22, 2023, the team led by Wensheng Wei from Peking University published a research paper titled "Unbiased interrogation of functional lysine residues in human proteome" in Molecular Cell. The study utilized adenine base editor to establish a strategy for unbiasedly probing functional amino acid residues at the genome scale, obtaining a map of functional lysine residues in the proteome through cell fitness screen.
Among the numerous amino acid residues in the human proteome, researchers first focused on lysine. L-lysine contains an ε-amino group, which is positively charged at physiological pH and plays numerous essential roles in protein functions. Moreover, lysine residues undergo a wide range of reversible PTMs (such as ubiquitination, acetylation, methylation). Lysine residues are encoded by the genetic codes of 5’-AAA and 5’-AAG, which can be mutated by ABEs to the codons of arginine, glutamic acid, or glycine (Figure 1).
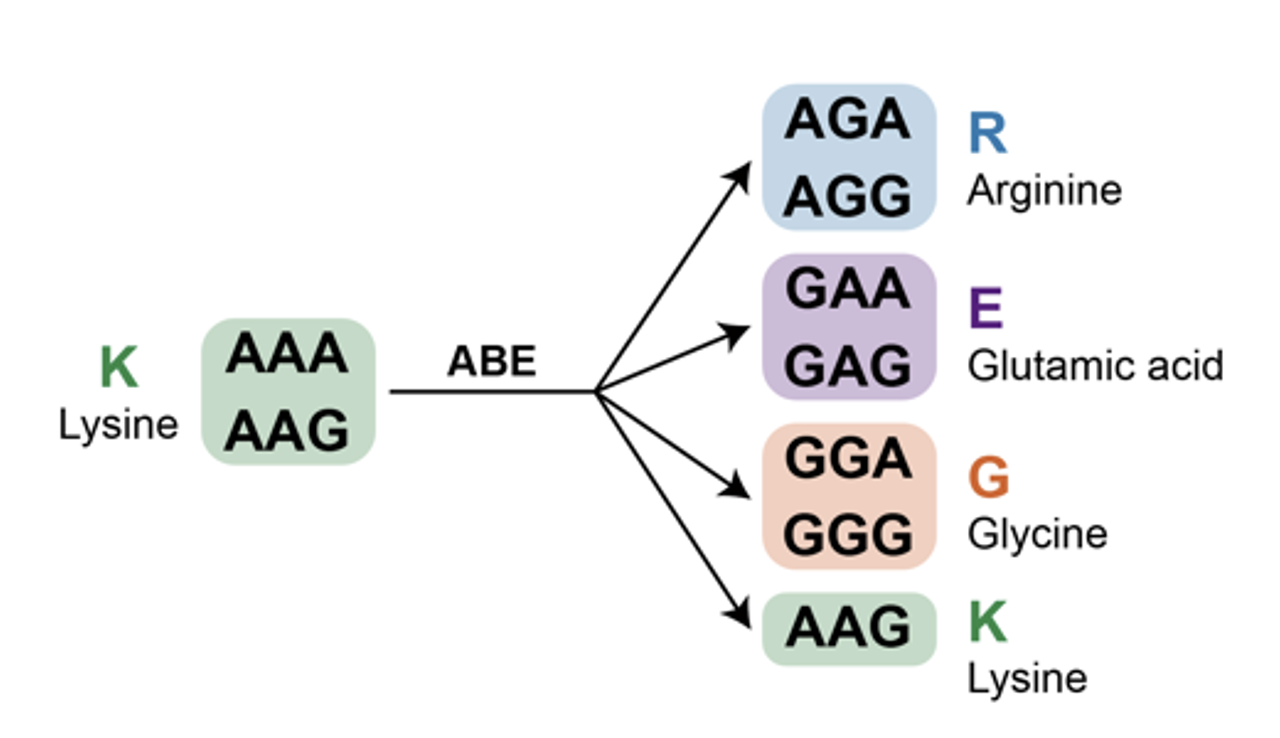
Figure 1. Possible editing consequences at lysine codons by ABE
By coupling adenine base editors and barcoded sgRNAs3,4, the researchers targeted 215,689 out of 611,267 (35%) lysine codons, involving 85% of the total protein-coding genes. Ultimately, they identified 1,572 lysine codons whose mutations perturb human cell fitness, with many of them implicated in cancer.
Based on the previous gene knockout screening results in the RPE1 cell line5, the researchers mapped the scores of lysine residues to their corresponding genes, creating a functional map of genes (proteins) and lysine residues (Figure 2). It is noteworthy that a large number of mutations in lysine residues led to cellular adaptability phenotypes different from those resulting from the corresponding gene knockout. Among these mutation sites, 805 sites showed inhibited cell survival after mutation, while the corresponding gene knockout did not affect cell survival or even promoted it. Through data mining of the International Cancer Genome Consortium (ICGC) database, the researchers identified several clinically significant lysine mutation sites, including reported sites such as TP53-K120, BRAF-K601, PTEN-K13. However, the functions of the majority of lysine mutation sites remain unknown.
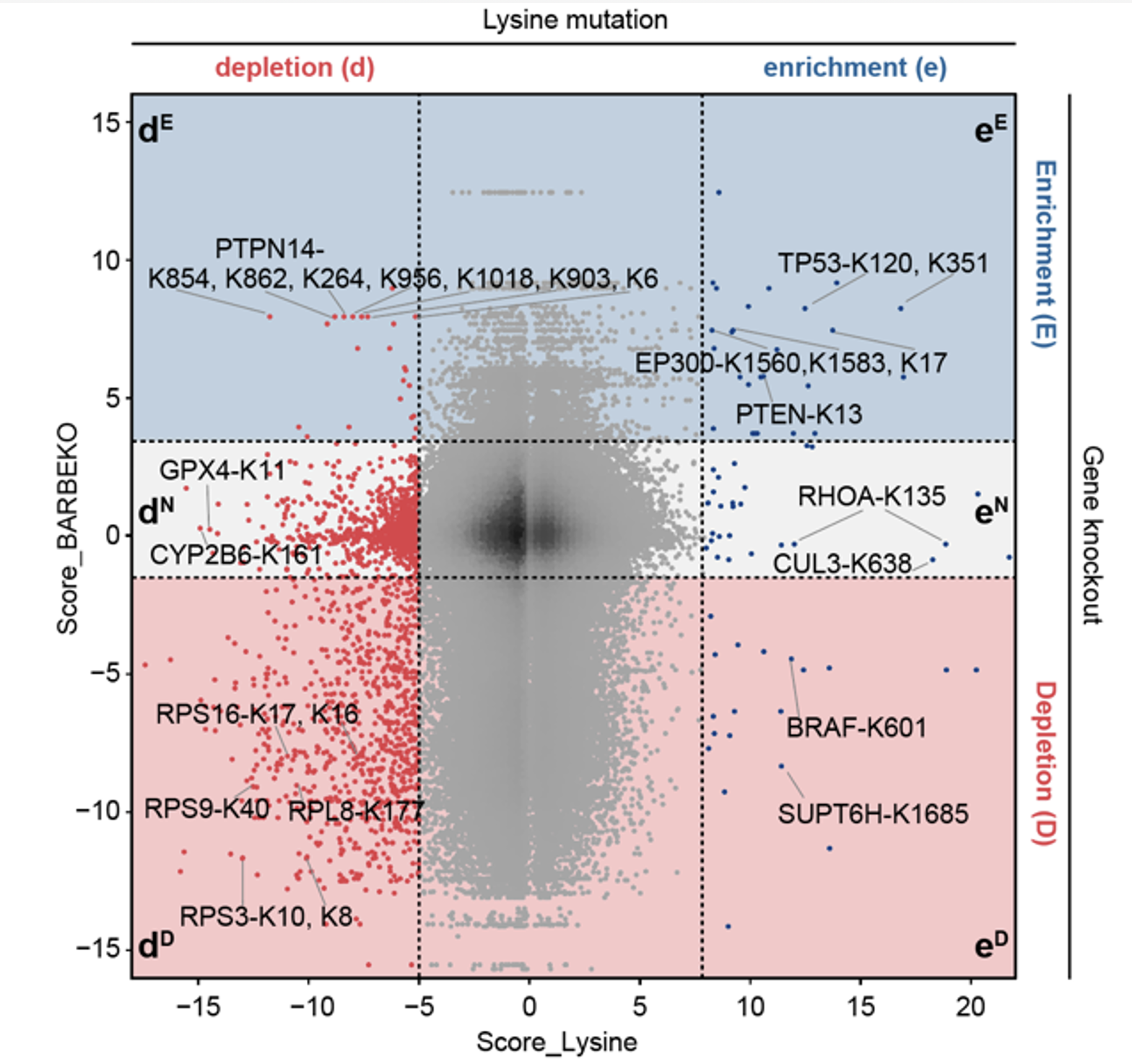
Figure 2. Scatter plot showing the distribution of FS values of lysine residues and their corresponding genes.
In the enriched lysine residues, the researchers identified a regulatory network centered around CUL3. CUL3 is an important member of the ubiquitin ligase complex (cullin-RING ligases, CRLs). Functional lysine residues were identified in CUL3, its adaptor protein, activating protein, and substrate protein (Figure 3). Using affinity purification-mass spectrometry and other methods, they found that CUL3-K638E significantly weakened the binding between CUL3 and the COP9 signalosome (CSN), leading to its sustained neddylation state and ultimately reducing its stability.
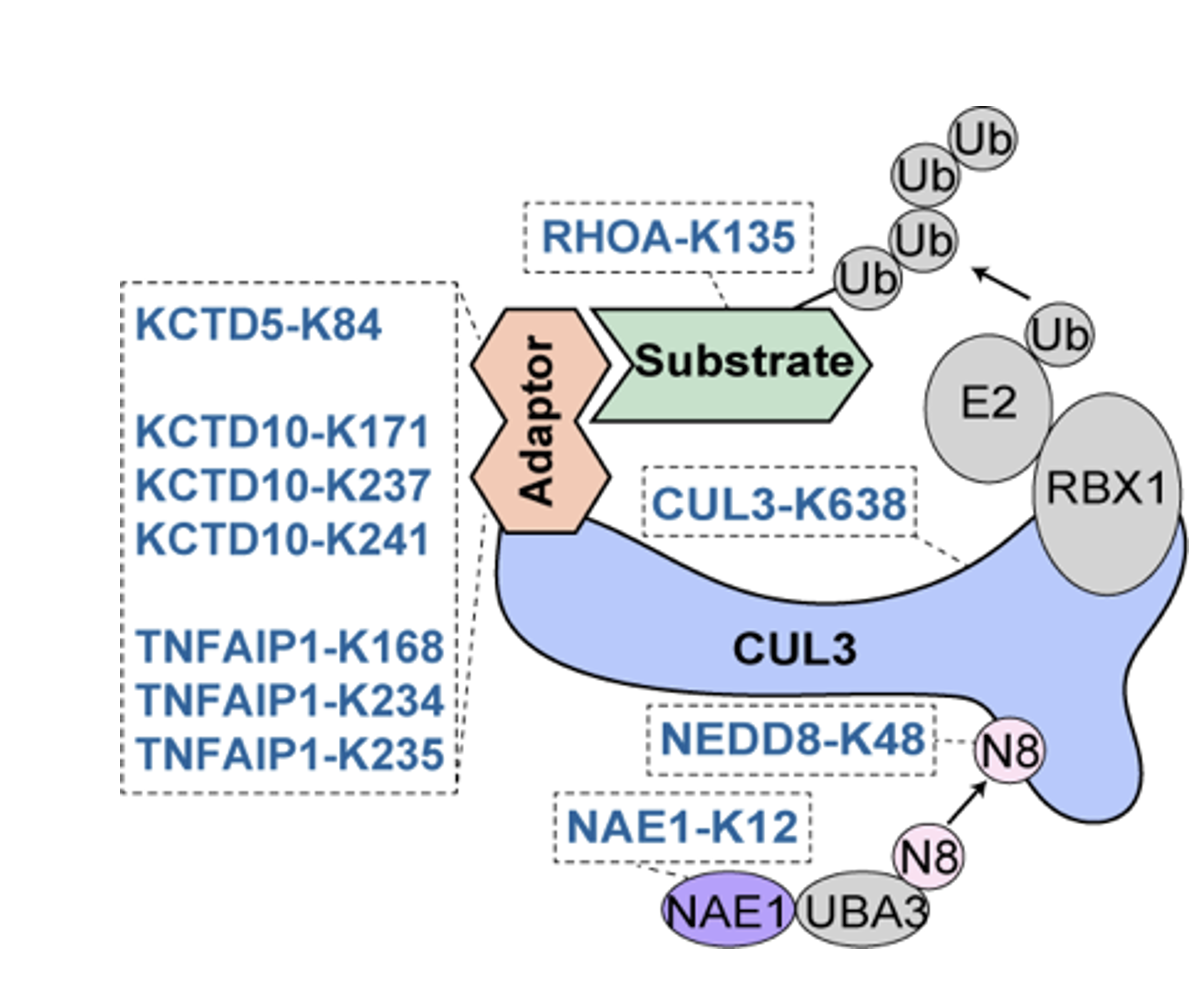
Figure 3 CUL3 CRLs complex
Ultimately, the researchers focused their attention on the K171 site of the adaptor protein KCTD10 in CUL3, which was found to be mutated (K171E) in a breast cancer patient. Through a series of experiments, the researchers confirmed that KCTD10-K171 can undergo acetylation modification. By regulating the protein stability of cell cycle proteins TPX2 and INCENP, it controls normal cell proliferation. The mutation of KCTD10-K171 may lead to the abnormal degradation of cell cycle proteins, resulting in excessive cell proliferation. This site may potentially serve as a potential biomarker in cancer diagnosis, prognosis, and therapy.
Overall, this study establishes a CRISPR strategy for unbiasedly probing functional amino acid residues at the genome scale. They systematically survey the functional lysine residues that affect cell fitness. Novel mechanisms that govern the cell cycle via lysine residues are unraveled, and clinically significant lysine mutations are identified. It offers a general strategy for interrogating genetic elements and provides functional insights into the human proteome (Figure 4).
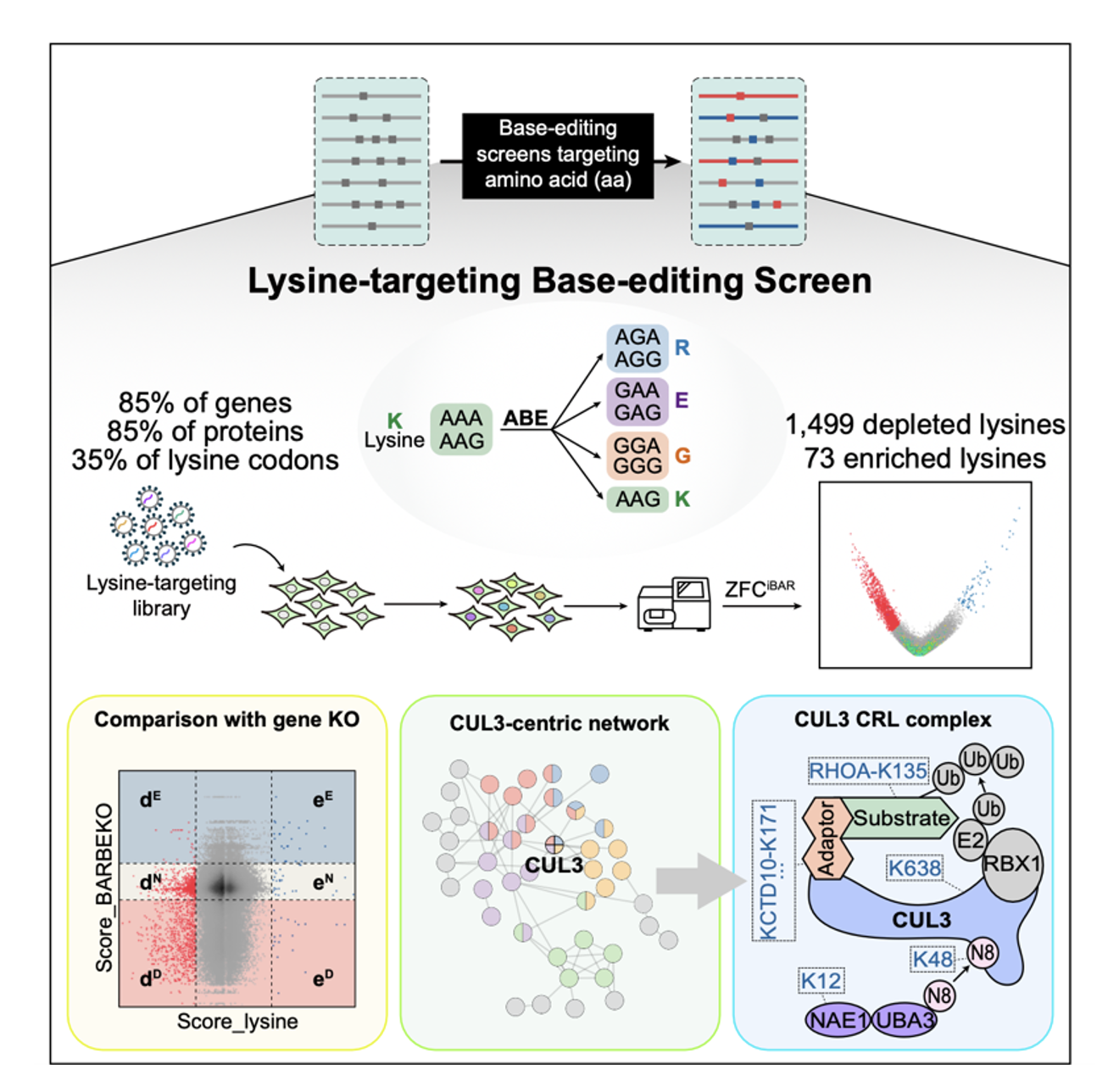
Figure 4. Summary
The research paper's co-first authors are Dr. Ying Bao, a postdoctoral researcher in the Wensheng Wei research group at Peking University, Qian Pan, a phD student, graduates Dr. Xu Ping and Dr. Zhiheng Liu. Professor Wensheng Wei and associate researcher Zhuo Zhou (now a researcher at the Institute of Systems Medicine, Chinese Academy of Medical Sciences /Suzhou Institute of Systems Medicine) are the corresponding authors of the paper. This project was funded by the National Key R&D Program of China, National Natural Science Foundation of China, the Beijing Advanced Innovation Center for Genomics at Peking University, the Peking-Tsinghua Center for Life Sciences, the Non-profit Central Research Institute Fund of the Chinese Academy of Medical Sciences, CAMS Innovation Fund for Medical Sciences, the State Key Laboratory Special Fund, the NCTIB Fund for R&D platform for Cell and Gene Therapy, and the Suzhou Municipal Key Laboratory.
Link: https://doi.org/10.1016/j.molcel.2023.10.033
Reference:
Komor, A.C., Kim, Y.B., Packer, M.S., Zuris, J.A., and Liu, D.R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. 10.1038/nature17946. Gaudelli, N.M., Komor, A.C., Rees, H.A., Packer, M.S., Badran, A.H., Bryson, D.I., and Liu, D.R. (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. 10.1038/nature24644. Koblan, L.W., Doman, J.L., Wilson, C., Levy, J.M., Tay, T., Newby, G.A., Maianti, J.P., Raguram, A., and Liu, D.R. (2018). Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846. 10.1038/nbt.4172. Zhu, S., Cao, Z., Liu, Z., He, Y., Wang, Y., Yuan, P., Li, W., Tian, F., Bao, Y., and Wei, W. (2019). Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol. 20. 10.1186/s13059-019-1628-0. Xu, P., Liu, Z., Liu, Y., Ma, H., Xu, Y., Bao, Y., Zhu, S., Cao, Z., Wu, Z., Zhou, Z., et al. (2021). Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 10.1038/s41587-021-00944-1.