NAR | High-Precision Haplotype Phasing Using Single-Sperm Long-Read Genome Sequencing
Haplotype phasing is the process of accurately determining the linkage relationships between various genetic polymorphisms on two homologous chromosomes within diploid cells. It is an essential component for achieving human reference genome assembly from telomere to telomere. Haplotype phasing aids in studying cis-interactions among different regulatory element genetic variations on the same chromosome. For example, it helps determine whether two heterozygous single nucleotide polymorphisms (SNPs) on different exons of the same gene are on the same homologous chromosome or on two separate homologous chromosomes. This is crucial for determining the functional state of the gene, whether it's in a homozygous mutant state (both alleles of the gene are inactive) or in a heterozygous mutant state (one allele is inactive, and the other is functional). Similarly, the phase relationship of two pairs of heterozygous SNPs in enhancer-promoter regions (or two different enhancers) of the same gene also needs to be determined through haplotype phasing.
Currently, the most common method for haplotype phasing involves high-throughput sequencing of both parental genomes for a specific individual to achieve haplotype phasing for the diploid genome of that individual. In cases where parental information is unavailable, local haplotype phasing of the genome can be accomplished using long reads, ultra-long reads, or linked-read sequencing. However, achieving whole chromosome-scale haplotype phasing (ranging from several million to several hundred million base pairs) requires additional techniques such as single chromosome sorting, Strand-seq, and Hi-C, which can resolve longer genetic polymorphism linkage information and provide haplotype phasing results on a whole chromosome scale. Nevertheless, these methods are time-consuming and costly. In contrast, direct whole-genome sequencing of individual gametes (haploid cells) is a more efficient and precise method for haplotype phasing.
The first single-sperm genome sequencing technology was developed ten years ago, and since then, it has been continuously optimized and improved, providing powerful technical support for research on meiosis, genomic instability in germ cells, and haplotype phasing. However, up to now, almost all single-sperm genome sequencing methods have been limited to second-generation sequencing platforms. This limitation is due to the obtained sequence lengths, which do not exceed 600 base pairs. Research on haplotype phasing is confined to the SNP level. Studies on genomic instability mostly focus on copy number variations (CNVs) and single nucleotide variations (SNVs). Identifying and phasing more important genetic polymorphisms on the genome, such as structural variations, especially those in the range of 50 base pairs to 10 kb pairs, is challenging.
On June 24, 2023, Tang Lab from Biomedical Pioneering Innovation Center (BIOPIC) and Beijing Advanced Innovation Center for Genomics (ICG) published a research paper in Nucleic Acids Research titled "Long-read-based single-sperm genome sequencing for chromosome-wide haplotype phasing of both SNPs and SVs."

This study introduced a novel approach for genome sequencing based on single-molecule sequencing (third-generation sequencing) platforms, specifically designed for single-sperm samples. The research also included the development of corresponding data analysis methodologies. High-quality single-sperm genome sequencing data obtained using this method enable precise identification of recombination events during sperm meiosis and accurate detection of structural variations within individual sperm cell genomes. Most importantly, single-sperm long-read genome sequencing achieves chromosome-wide haplotype phasing, covering both single nucleotide polymorphisms (SNPs) and structural variations (SVs) across the entire chromosome (Fig 1).
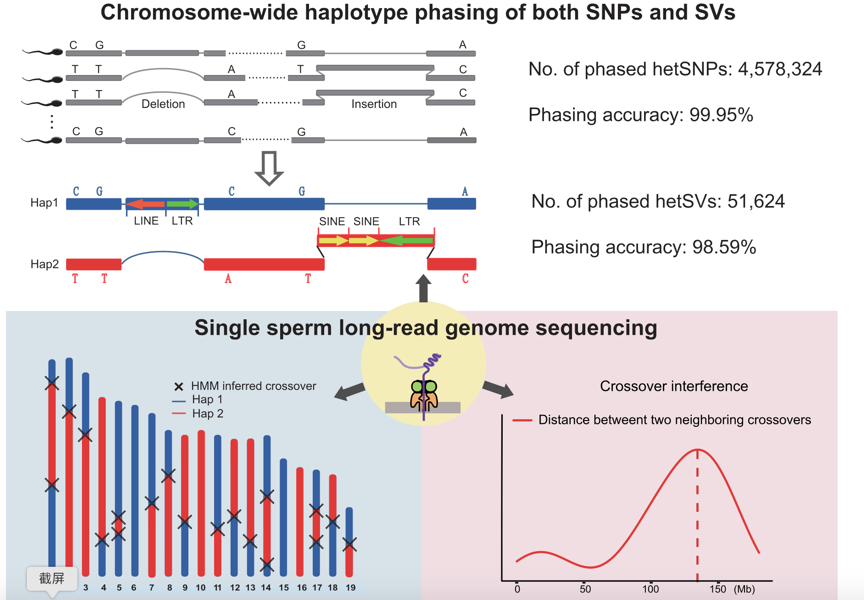
Figure 1. Single sperm genome sequencing method and its application based on long-read sequencing platform.
1. For the first time, a high-throughput single-sperm long-read genome sequencing method has been developed based on single-molecule sequencing platforms (suitable for both ONT and PacBio single-molecule sequencing platforms). In this study, 24 different barcode were designed for the Tn5 enzyme, and, in combination with the 96 barcode used in subsequent amplification primers, double-barcode labeling of individual sperm was achieved, allowing for a maximum throughput of up to 2304 single cells in a single sequencing run. Subsequently, the genomes of 24 sperm with different Tn5 transposon labels were pooled together for genome amplification, improving the evenness of amplification. Rigorous cross-contamination experiments were conducted to validate the low cross-contamination rate and reliability of the method. In the end, genome sequencing data were obtained from a total of 1,573 sperm cells from B6D2F1/Crl [BDF1] male hybrid mouse (samples containing multiple sperm cells or samples with genome coverage below 1% were filtered out). With a sequencing depth of 0.1×, individual sperm genomes achieved coverage ranging from 1% to 25.5% (with a median genome coverage of 4.9%), and the average sequencing read length was 5.5kb (Fig 2).
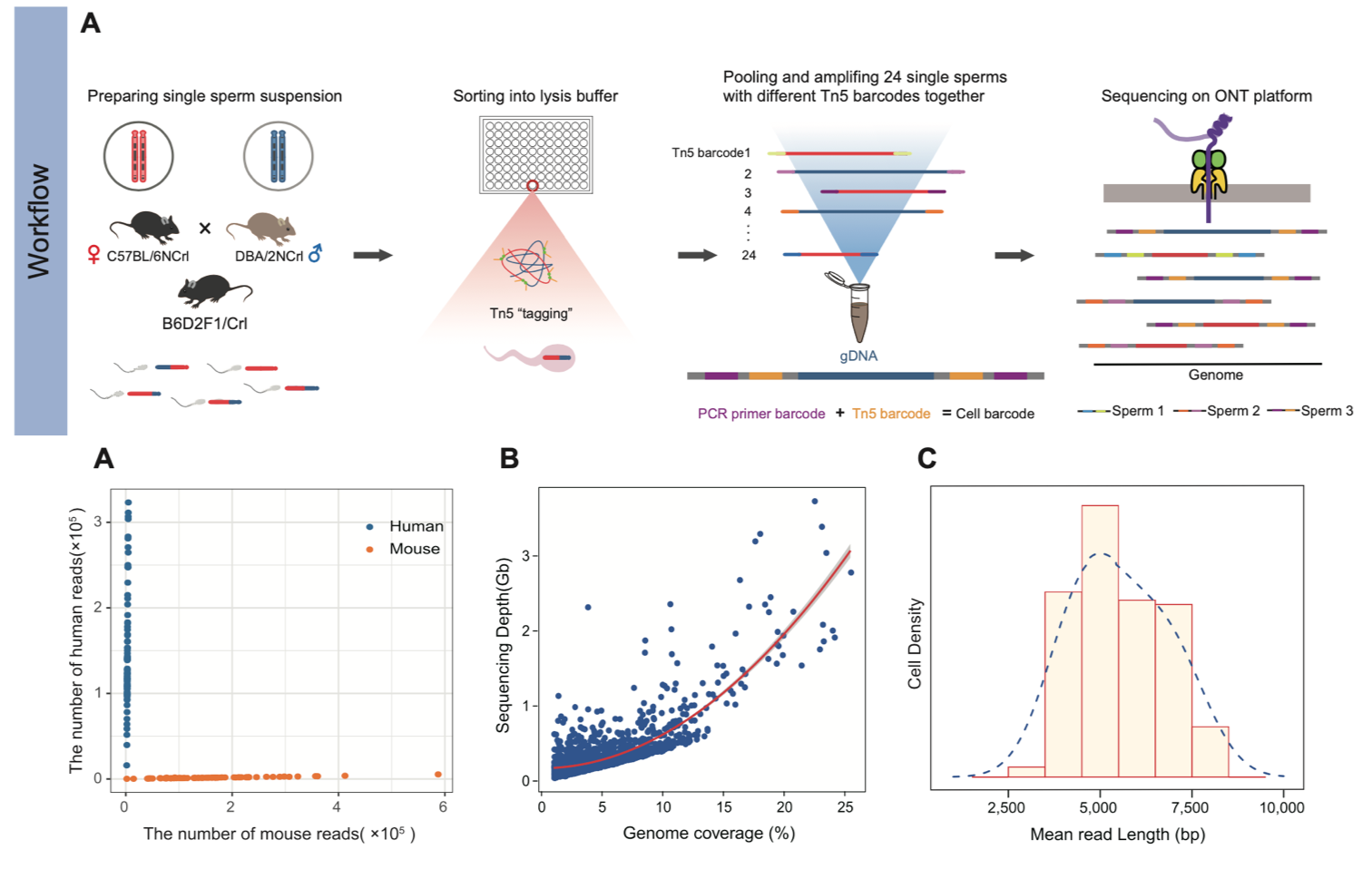
Figure 2. Experimental workflow and data quality control for single sperm genome sequencing method based on long-read sequencing platform.
2. Accurate identification of meiotic crossover recombination events and aneuploidy events in each sperm cell. This study employed a Hidden Markov Model (HMM) to determine the meiotic crossover recombination sites within individual sperm. In the end, 17,445 autosomal crossover recombination events were identified in the 1,573 sperm samples. Each sperm experienced 4 to 27 crossover events (with an average of 12 events per sperm). Additionally, crossover interference phenomena were detected in the single-sperm genome sequencing data. Furthermore, 29 entire chromosome loss events were identified in 23 sperm cells, including 15 on autosomes and 14 on sex chromosomes. Additionally, 4 sperm cells exhibited an increase in autosomal copy number (Fig 3).
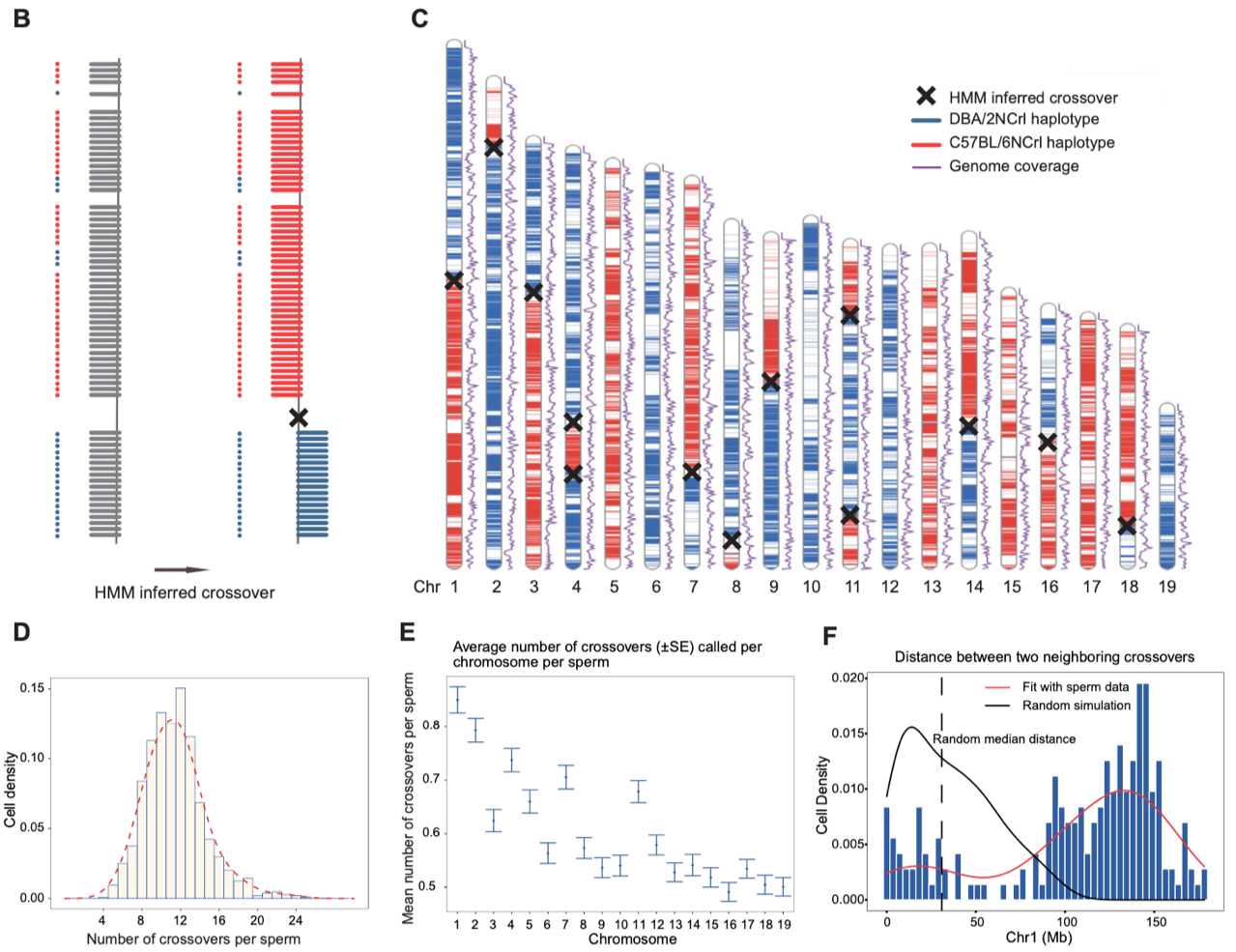
Figure 3. Identification of crossover events in single sperm.
3. Precise identification of structural variations (SVs) within individual sperm cells. In this study, the structural variations identified in bulk ONT single-molecule sequencing data from the parental mouse (serving as the gold standard) were used for evaluation. A total of 57,116 and 5,901 structural variations were identified in the genomes of DBA/2NCrl and C57BL/6NCrl mouse, respectively. This served to assess the accuracy of structural variation detection in single-sperm genome sequencing data. The study found that the detection accuracy of structural variation events supported by six or more sperm cells could reach 90%, while those supported by three sperm cells achieved the highest F1 score (78%). Additionally, 70 structural variations involving repetitive elements were selected for PCR experiments, and the validation accuracy reached approximately 90% (Fig 4).
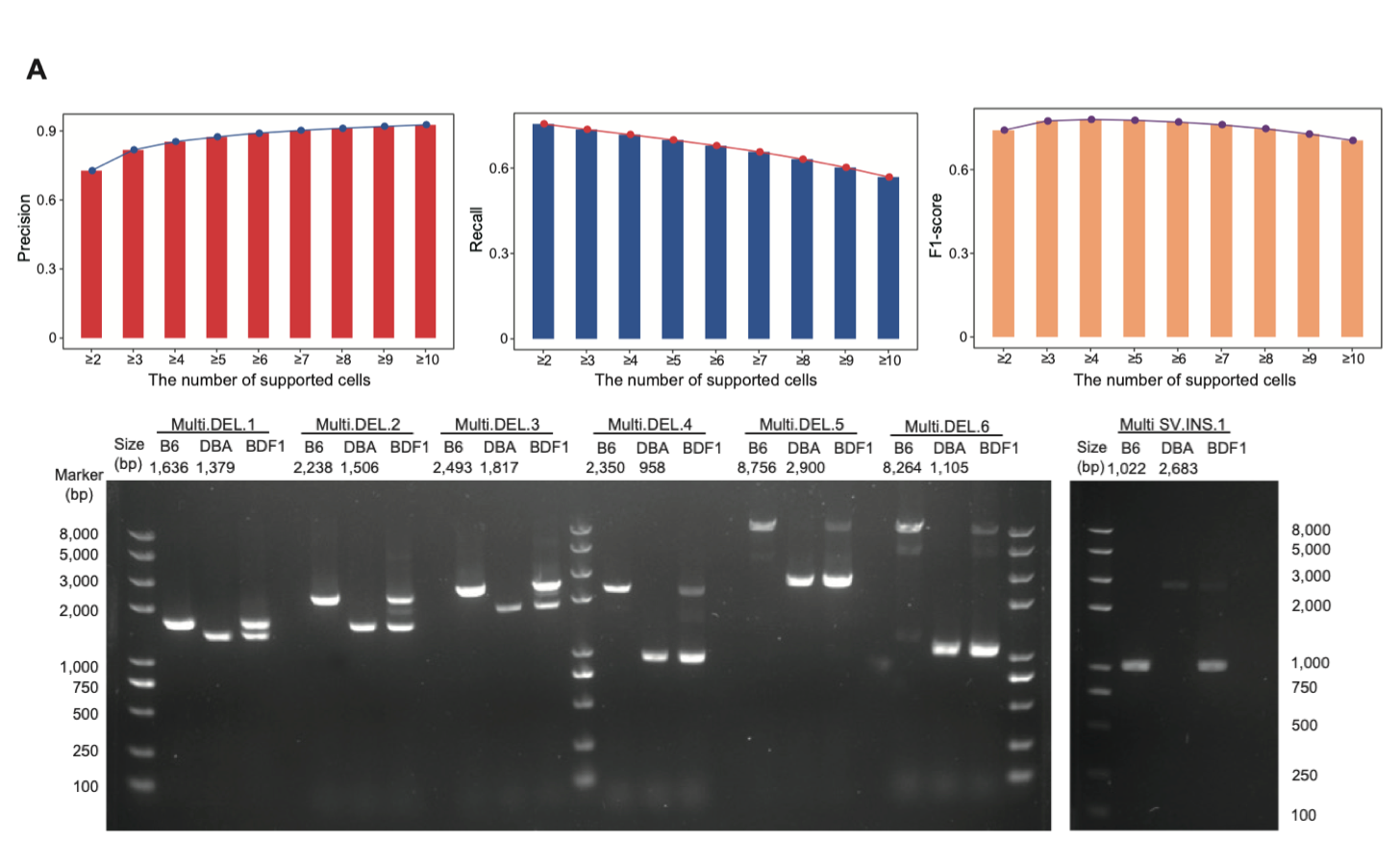
Figure 4. Identification of genomic structural variations in single sperm.
4. High-precision haplotype phasing of genetic polymorphisms, including SNPs and structural variations, at the whole chromosome scale has been achieved. This study introduced a novel chromosome-scale haplotype phasing workflow. In this workflow, a total of 54,712 heterozygous structural variations were identified, and 94.36% of these heterozygous structural variations could be phased with this technology, achieving a phasing accuracy of 98.59%. Moreover, a total of 4,664,507 heterozygous SNPs were identified, and 98.15% of these heterozygous SNPs could be phased using this technology, with a phasing accuracy of 99.95%. Meanwhile, the length distribution of successfully phased structural variations revealed peaks at around 190bp and 6kb, corresponding to two abundant classes of repetitive elements in the mammalian genome: SINE and LINE (Fig 5).
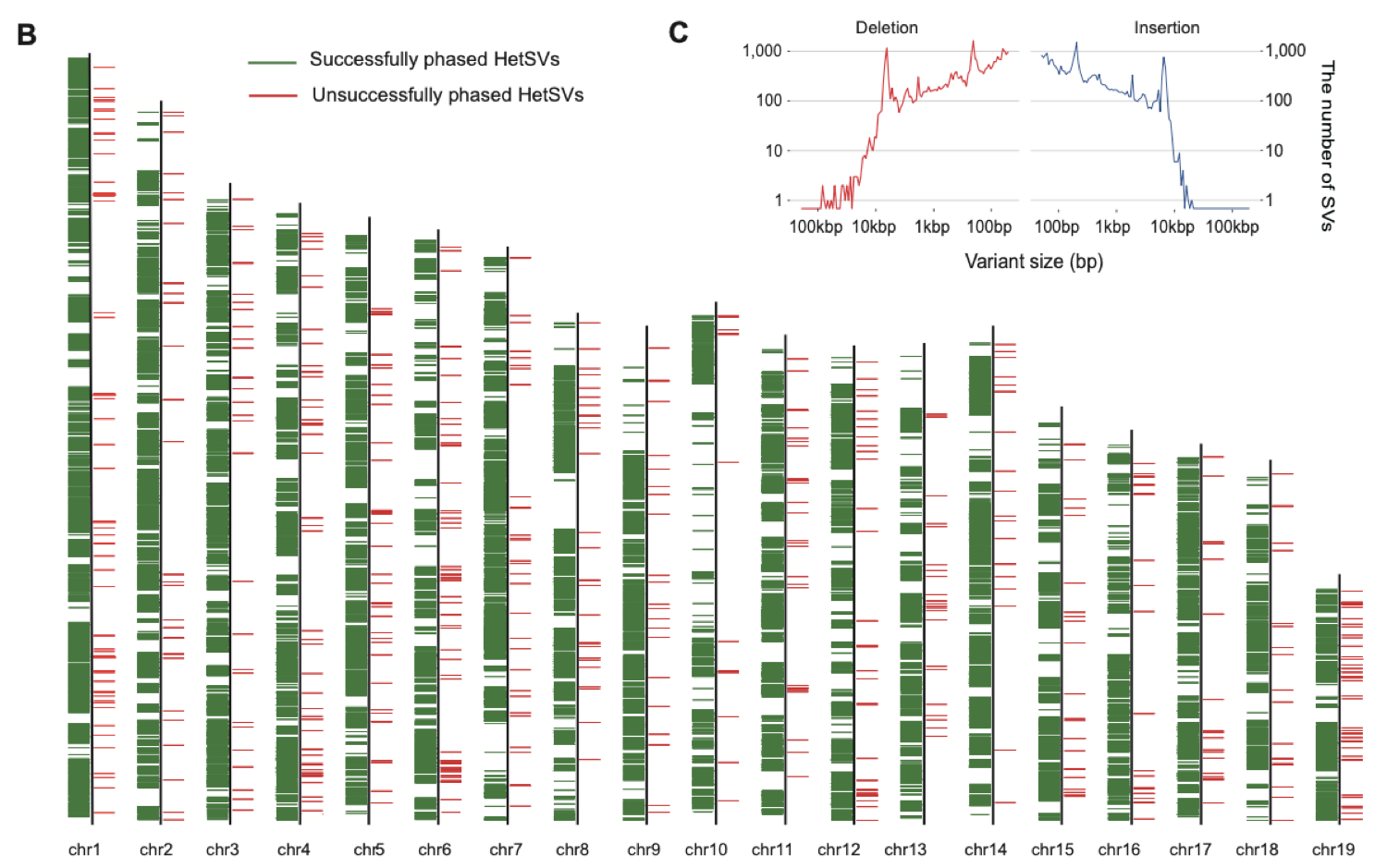
Figure 5. Chromosome-scale haplotype phasing using single sperm genome sequencing data.
5. Accurate identification and classification of structural variations containing repetitive elements. Among the 36,271 successfully phased heterozygous structural variations with a length greater than 100bp, 25,664 of them included repetitive elements. Specifically, 29.3% were covered by individual LINEs, 17.0% were covered by individual SINEs, 16.2% were covered by individual LTRs, 12.4% were covered by tandem repeats, and 24.9% were covered by combinations of multiple repetitive elements. Additionally, 3,190 tandem repeat sequences were identified with specific amplifications between haplotypes. To visualize the structural and sequence composition differences at these tandem repeat loci between the B6 and DBA haplotypes, this study employed k-mer frequency information (Fig 6).
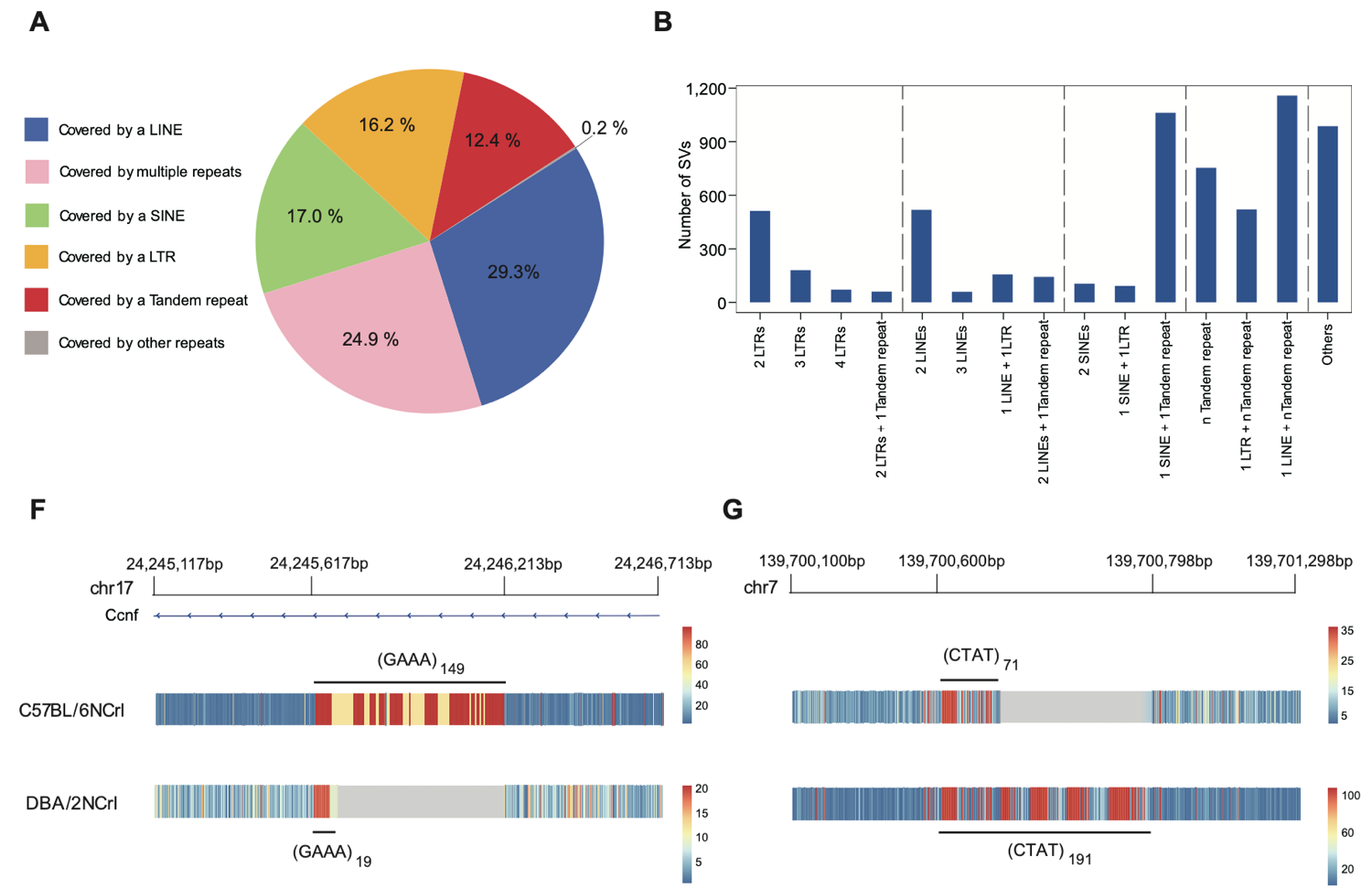
Figure 6. Identification and visualization of genomic structural variations involving repetitive elements.
This study has several potential applications: Firstly, similar to the Strand-seq technique, this method can be combined with long-read genome sequencing of bulk to achieve high-precision haplotype assembly. Alternatively, by conducting high-depth long-read genome sequencing on hundreds of individual sperm cells, it can directly accomplish haplotype assembly. Secondly, this method can be applied to the study of individual human sperm cells. By identifying genome structural variations (including repetitive elements) in infertile male reproductive cells, it can provide new insights for research on male infertility and genetic diseases. The research results indicate that achieving high-precision whole-chromosome-scale haplotype phasing only requires a minimum of 100 sperm cells. This significantly reduces the cost of haplotype phasing and opens up new avenues for building a comprehensive human pan-genome.
The first co-authors of this paper are Haoling Xie (Ph.D.) from Biomedical Pioneering Innovation Center of Peking University, Wen Li (Ph.D. candidate) from the Academy for Advanced Interdisciplinary Studies of Peking University, and Yuqing Guo (Ph.D. candidate) from the School of Life Sciences of Peking University. Professor Fuchou Tang of Beijing Advanced Innovation Center for Genomics, Biomedical Pioneering Innovation Center is the corresponding author. This research project was supported by Peking University-Tsinghua University Joint Center for Life Sciences, Beijing Changping Laboratory, and the Beijing Municipal Science and Technology Commission.