Cell Research | scNanoCOOL-seq: a long-read single-cell sequencing method for multi-omics profiling within individual cells
On June 30th, 2023, Fuchou Tang’s Lab in Biomedical Pioneering Innovation Center (BIOPIC) and Beijing Advanced Innovation Center for Genomics (ICG) in Peking University published an article titled “scNanoCOOL-seq: a long-read single-cell sequencing method for multi-omics profiling within individual cells” on Cell Research. This study reported a novel single-cell multi-omics sequencing technique based on the third-generation sequencing (TGS) platform, which allowed for concurrent analysis of genome (CNV), DNA methylome, chromatin accessibility, and transcriptome in long reads of individual cells.

DNA methylation plays a crucial role in biological processes such as biological development and tumorigenesis. Similar to scCOOL-seq previously reported by the group, scNanoCOOL-seq was based on the NOMe-seq method which used the DNA methyltransferase M.CviPI's to methylate GpC sites within open chromatin regions. Different from scCOOL-seq, the scNanoCOOL-seq technology got DNA amplicons about 900 bp in length and used Oxford Nanopore platform for sequencing (Figure 1). This allowed researchers to detect DNA methylation and chromatin status over a relatively long genomic region.

Figure 1. Flowchart of scNanoCOOL-seq
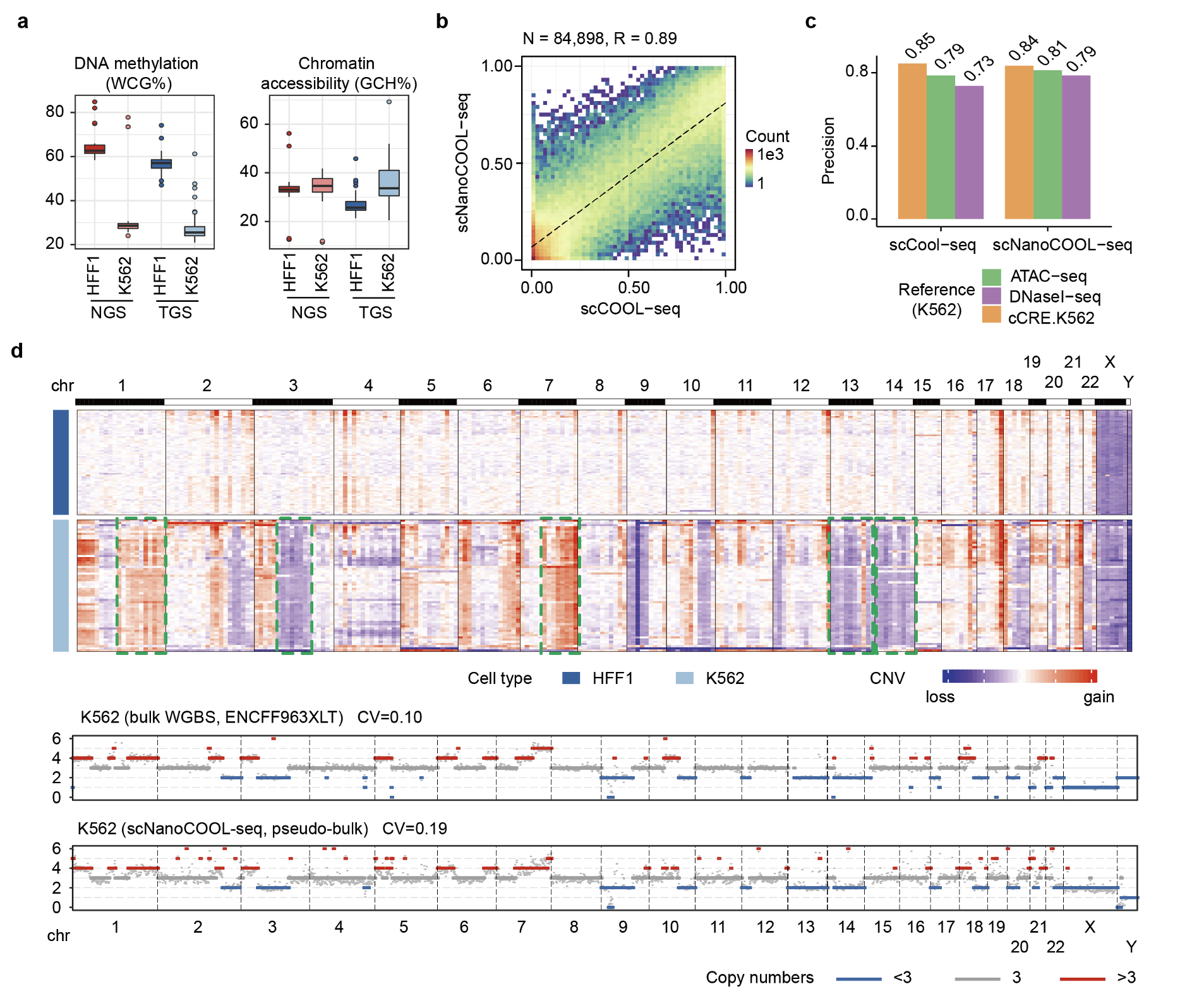
Figure 2. Validation of scNanoCOOL-seq’s detection accuracy
Several key conclusions emerge from this work:
1. scNanoCOOL-seq can accurately detect epigenetic features such as DNA methylation and chromatin accessibility in single cells with single-molecule resolution.
The researchers first showed that scNanoCOOL-seq accurately detected DNA methylome, chromatin accessibility, and genome (CNV) at a single-cell level (Figure 2). Compared to scCOOL-seq, scNanoCOOL-seq achieved much longer read length. CpG islands (CGIs) had a median length of 575 bp in human genome and cannot be fully covered by NGS sequencing. The researchers showed that scNanoCOOL-seq covered hundreds of full length CpG islands in single cells, which helped better understanding regulatory effects of DNA methylation on gene expression (Figure 3a, b). Long read length of scNanoCOOL-seq also enabled detecting the epigenetic status of complex structural variations (SVs), such as translocations. The researchers found that scNanoCOOL-seq accurately detected the DNA methylation and chromatin accessibility of the BCR-ABL1 fusion gene caused by translocation and their corresponding wild-type alleles in K562 cells at single-molecule resolution (Fig. 3c). The scNanoCOOL-seq also accurately detected allele-specific DNA methylation patterns in imprinted regulatory regions of imprinted genes (Figure 3d).
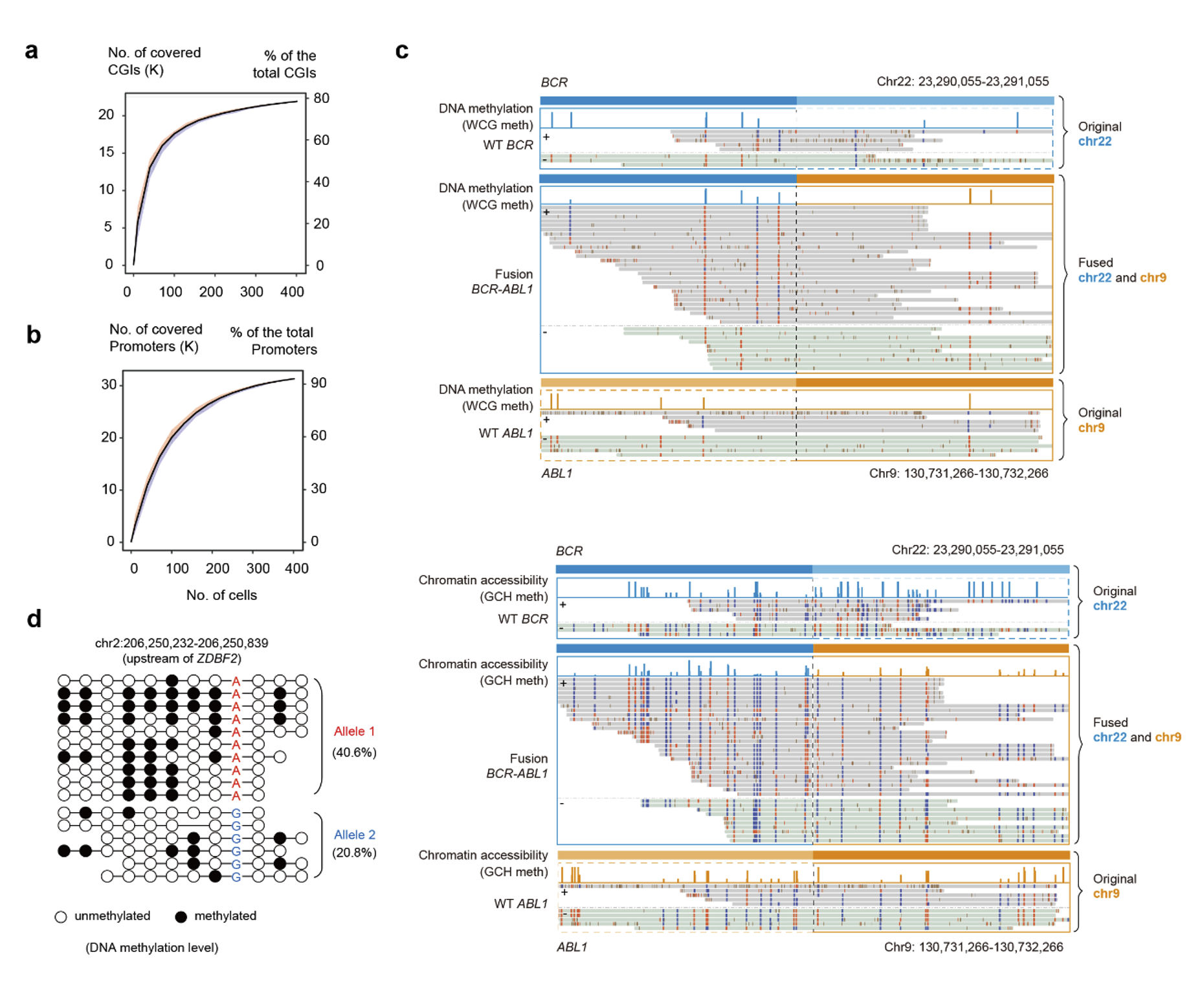
Figure 3. Epigenetic signatures of full-length CpG islands, gene promoters, chromatin translocation events, and allele-specific DNA methylation detected by scNanoCOOL-seq
2. scNanoCOOL-seq can accurately detect the dynamic changes and heterogeneity of DNA methylation and chromatin accessibility in single cells
5-Azacytosine (5-aza) is a common DNA methylation inhibitor. The researchers used 5-aza-treated K562 cells as a biological model to explore the ability of scNanoCOOL-seq to detect the dynamic changes and heterogeneity in DNA methylation and chromatin accessibility (Figure 4a). The results showed that the DNA methylation levels continue decreasing after treatment with 5-aza for 5 days (Figure 4b, c). The cells showed increased heterogeneity after the treatment (Figure 4d).
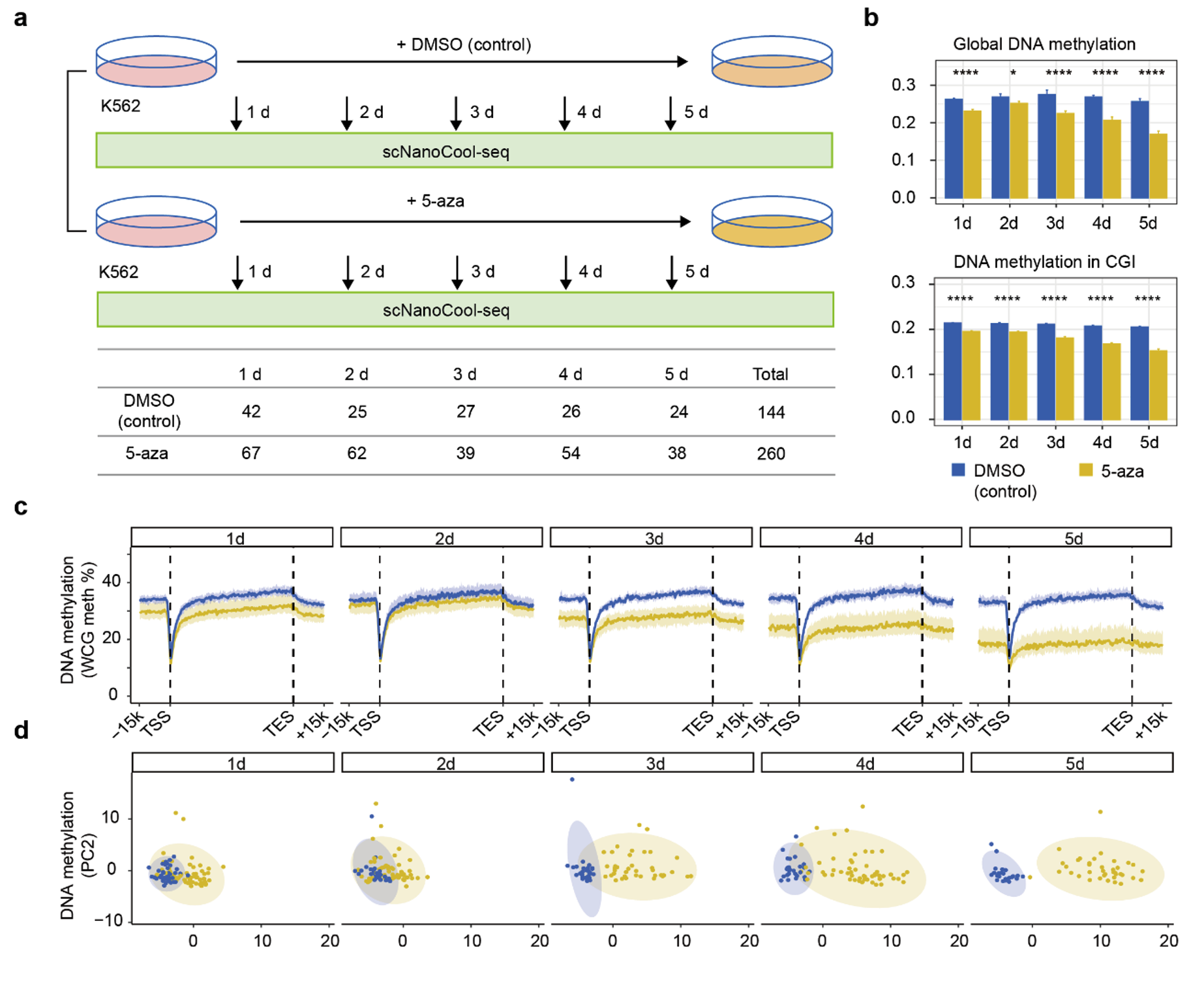
Figure 4. scNanoCOOL-seq’s detection on the dynamics and heterogeneity of K562 cells in response to demethylation drug 5-aza
3. scNanoCOOL-seq can accurately characterize cell lineage-specific epigenome during mouse blastocyst development
Preimplantation embryos of mammals undergo critical epigenetic reprogramming and two cell fate decisions. The first cell fate decision occurs when the early blastocyst differentiates into the pluripotent inner cell mass (ICM) and trophectoderm (TE). The second cell fate decision occurs when the ICM cells at the late blastocyst stage further differentiate into epiblast (Epi) and primitive endoderm (PrE) (Figure 5a). The researchers used scNanoCOOL-seq to study mouse blastocyst-stage embryos (Figure 5b). They found that i) in late blastocysts (E4.5), the DNA methylation level of the gene body region in PrE cells (22.8%) was higher than that in Epi cells (18.8%), while the lowest was found in TE cells (15.1%); ii) in the early female (XX) blastocyst, imprinted X chromosome inactivation (iXCI) seemed not completed until the late blastocyst stage, and some Epi cells of late female blastocysts seemed to be reactivated (Figure 5c); iii) compared to the Epi cells, the maternal genomic DNA methylation in the late TE cells had a stronger strand bias while a lower expression level of DNMT1, indicating that the TE cells had gone through a more intense passive DNA demethylation (DNA methylation was not completely maintained on the nascent DNA strand during DNA replication) (Figure 5d).
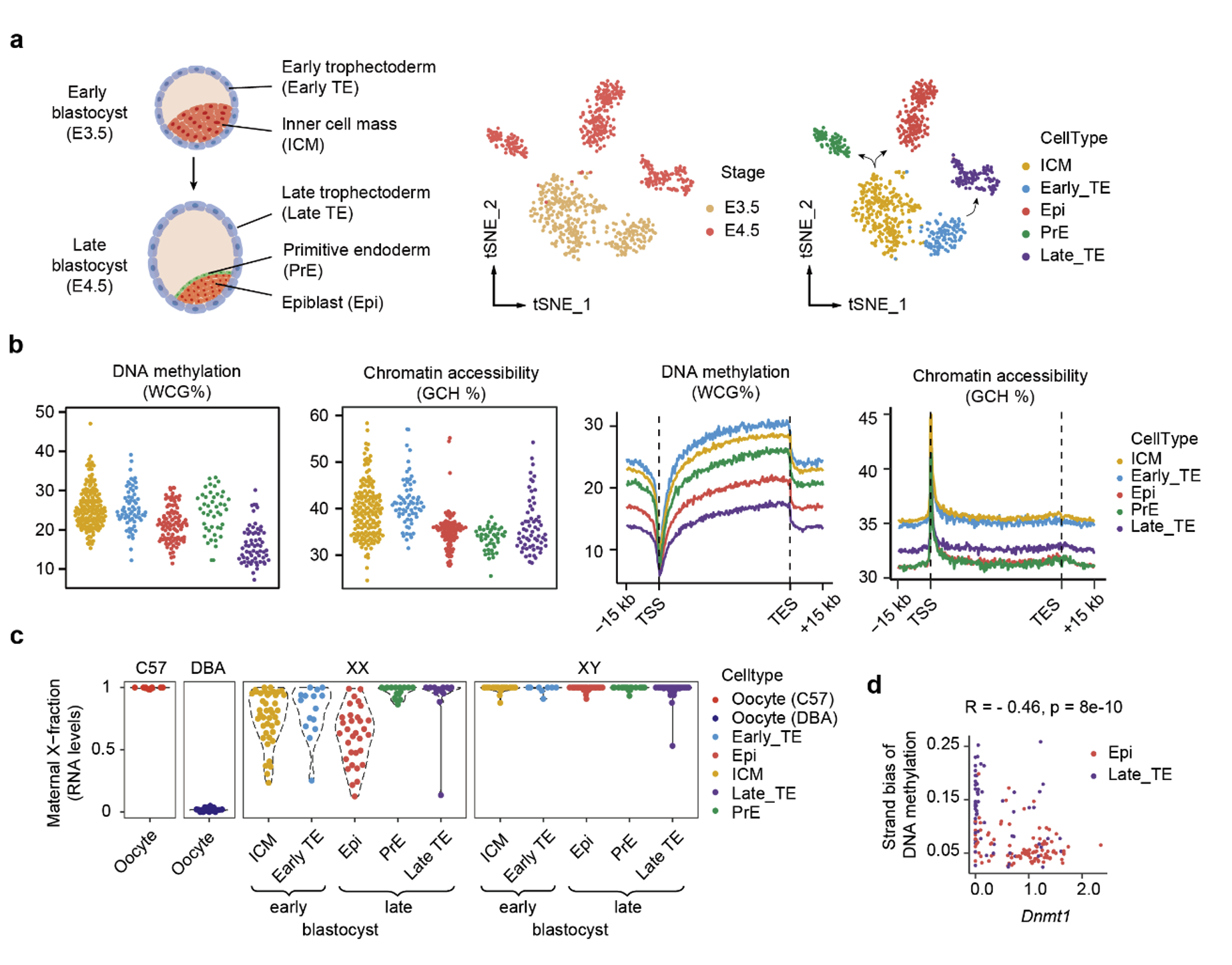
Figure 5. Identification of different lineages, detection of epigenomic dynamics, allele-specific transcriptome, and strand-specific epigenome in mouse blastocysts by scNanoCOOL-seq
In summary, scNanoCOOL-seq is a single-cell multi-omics sequencing method based on single-molecule sequencing platform. It can simultaneously analyze the genome (CNV), DNA methylome, chromatin accessibility, and transcriptome sequencing in an individual cell. Taking advantage of the long-read sequencing, scNanoCOOL-seq can detect multiple epigenetic features of full-length CGIs and gene promoter regions as well as the interactions between different omics levels. In addition, scNanoCOOL-seq can also detect epigenetic dynamics caused by complex genome structural variation events such as chromosomal translocations at single-molecule resolution. It can also detect allele-specific epigenetic features and DNA strands in single cells.
Ph.D. candidate Jianli Lin and Ph.D. candidate Xiaohui Xue from Academy for Advanced Interdisciplinary Studies (AAIS) of Peking University are the co-first authors of the paper. Prof. Fuchou Tang is the corresponding author. This work was supported by the Basic Science Center Projects of the National Natural Science Foundation of China and the Science and Technology Innovation “2030-Brain Science and Brain-like Research” major project.
Article link: https://www.nature.com/articles/s41422-023-00873-5