National Science Review∣Integrative single-cell multiomics analyses dissect molecular signatures of intratumoral heterogeneities and differentiation states of human gastric cancer
Gastric cancer is a highly prevalent and deadly malignancy characterized by strong tumor heterogeneity, often presenting with diverse histopathological subtypes clinically. According to the latest WHO classification of digestive system tumors, gastric cancer encompasses various pathological types, including tubular adenocarcinoma, papillary adenocarcinoma, poorly cohesive carcinoma, signet-ring cell carcinoma, neuroendocrine carcinoma, and hepatoid adenocarcinoma. Among them, mixed-type gastric cancer (mGC) is a typical representative with high intratumor heterogeneity. mGC contains at least two or more pathological components within the same tumor, making it more prone to recurrence and metastasis. However, the current role of gastric cancer classification based on histopathology in guiding clinical treatment remains limited, primarily due to inadequate understanding of the molecular characteristics associated with different pathological subtypes of gastric cancer. This calls for further investigation in this direction.
On April 12, 2023, the Tang Fuchou Research Group at Peking University's Beijing Biomedical Pioneering Innovation Center (BIOPIC) and the Center for Life Sciences (CLS) in collaboration with other researchers, published a research paper titled "Integrative single-cell multiomics analyses dissect molecular signatures of intratumoral heterogeneities and differentiation states of human gastric cancer" on National Science Review.

In this study, researchers utilized radical surgical resection specimens of gastric cancer and collected paired normal adjacent tissues (NATs), primary tumors, and lymph node metastases. Each tumor site was multi-regional sampled (Fig. 1). In total, 85 samples from 14 gastric cancer patients were profiled using the optimized single-cell multiomics sequencing (scTrio-seq3), which could analyze genome (somatic copy number alterations), DNA methylation, chromatin accessibility, and transcriptome. The researchers then performed systematic and in-depth integration of the data, incorporating the histopathological classification results from the corresponding sampling regions. They accurately depicted a single-cell DNA methylation atlas of gastric cancer, revealing key molecular features at multi-omics levels and immune microenvironment characteristics of cancer cells with different differentiation states in mGC. This study provides new theoretical foundations for the clinical diagnosis and treatment of gastric cancer.
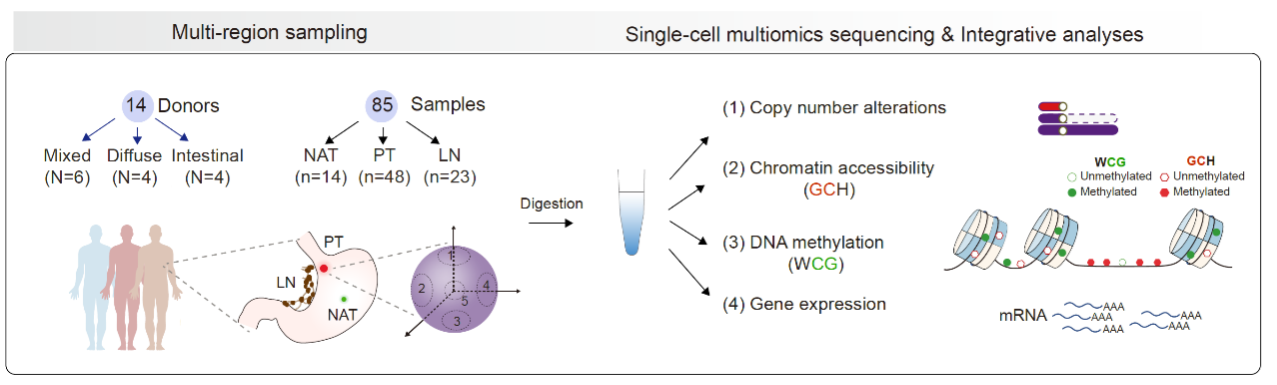
Fig. 1 Schematic illustration of the study workflow
The main findings of this study are as follows:
Revealing common patterns of abnormal gene expression in gastric cancer cells among different patients
The study identified 334 genes that were commonly upregulated and 82 genes that were commonly downregulated in the cancer cells of the 14 gastric cancer patients. Through systematic comparisons with normal gastric epithelial cells from gastric cancer NATs and normal intestinal epithelial cells from colorectal cancer NATs, they found that 26 genes among the upregulated genes in gastric cancer were genes specifically highly expressed in intestinal epithelial cells (Fig. 2). These genes include TFF3, CLDN3, CLDN4, CLDN7, TMEM176A, TMEM176B, and MUC13. This phenomenon was observed not only in the intestinal-type gastric cancer but also in the diffuse-type and mixed-type gastric cancer. In contrast, several genes that are specifically expressed in normal gastric epithelial cells and involved in normal gastric physiological functions were consistently and significantly downregulated in gastric cancer cells. These genes include PGA3, PGA5, PGC, LIPF, GKN1, GKN2, MUC1, MUC5AC, and MUC6. These findings suggest that during the tumorigenesis of gastric cancer, the cancer cells undergo dedifferentiation and lose some of the gene expression characteristics and normal physiological functions of normal gastric epithelium. Simultaneously, due to increased plasticity of these cancer cells, they aberrantly express dozens of genes that are normally highly expressed in intestinal epithelium. As the gastric epithelium and intestinal epithelium derive from common precursor cells in the digestive tract during development, the abnormal expression of certain intestinal epithelium-specific genes in gastric cancer cells indicates that while these cells lose some differentiation characteristics of gastric epithelium, they acquire a high degree of plasticity. This provides a new research perspective for understanding the characteristics of gastric cancer, such as its propensity for recurrence and drug resistance.
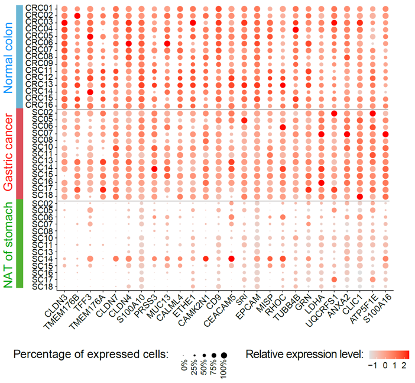
Fig. 2 26 genes highly expressed in normal colon epithelium and upregulated in gastric cancer cells.
Through integrated analysis of single-cell epigenomic data, they discovered that aberrant changes in chromatin accessibility and DNA methylation levels in gene promoter regions may explain the abnormal gene expression in gastric cancer cells (Fig. 3). This finding provides important clues for understanding the epigenetic regulation of gene expression abnormalities during the gastric cancer development.
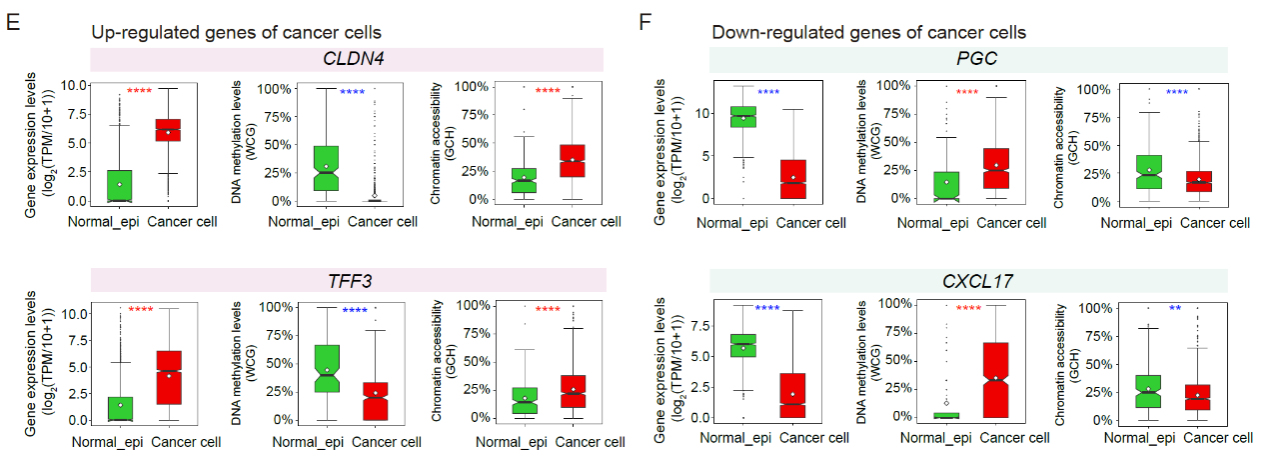
Fig. 3 he gene expression levels, DNA methylation levels and chromatin accessibility levels of related promoter regions of representative genes.
Single-cell DNA methylation alteration map of cancer cells of gastric cancers
The study systematically depicted the single-cell DNA methylation atlas of gastric cancer and found that gastric cancer cells underwent extensive genome-wide DNA demethylation during tumorigenesis. Particularly, there was a greater extent of DNA demethylation in genomic regions containing repetitive elements such as LINE, SINE, LTR, and Satellite. This DNA demethylation exhibited strong heterogeneity both between different patients and between cancer cells within the same patient. Compared to colorectal cancer, gastric cancer displayed a greater degree of genome-wide demethylation. Notably, the DNA methylation levels of LINE-1 elements in the gastric cancer genome were dramatically decreased, accompanied by abnormal upregulation of the ORF1p protein encoded by LINE-1. This finding suggests that some retrotransposons, particularly LINE-1, are likely to be abnormally activated for transcription and transposition in gastric cancer cells, thereby compromising genome integrity.
This study identified several genes (promoter regions) with either gastric cancer-specific hypermethylation or hypomethylation that were shared among multiple patients (Fig. 4). These genes could serve as candidate DNA methylation biomarkers for gastric cancer, such as the promoter regions of TMEM240 and HAGLROS, which exhibited abnormal hypermethylation, and the promoter regions of TRPM2-AS and HRH1, which showed abnormal hypomethylation. This discovery provides a theoretical basis for the clinical diagnosis of gastric cancer.
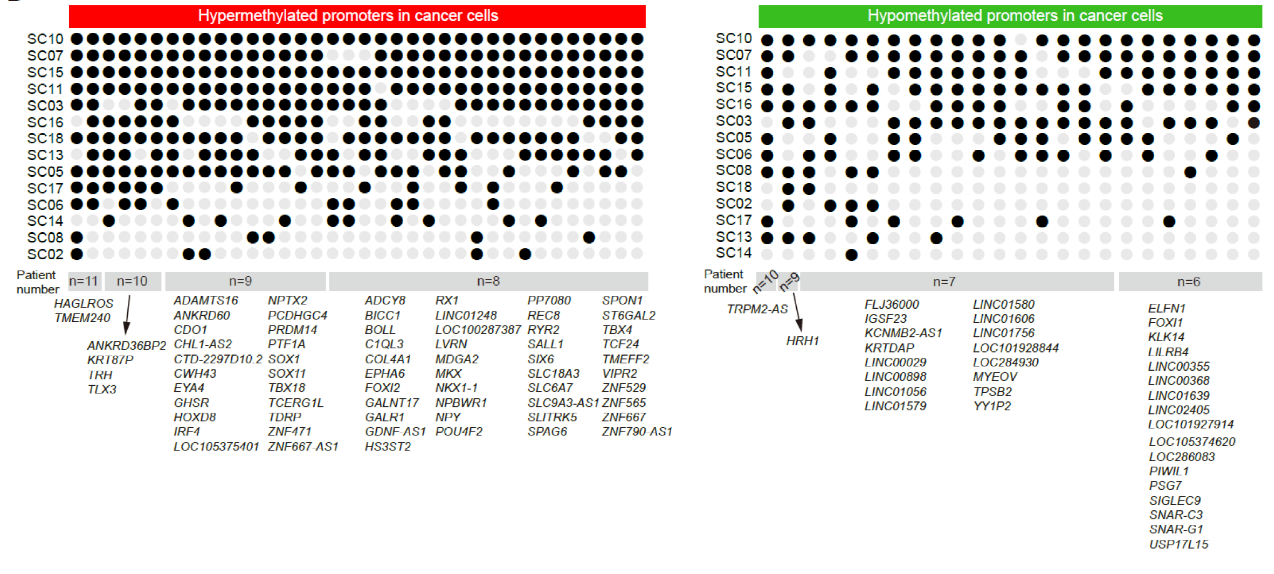
Fig. 4 Abnormally methylated genes (promoter regions) in gastric cancer cells shared by multiple gastric cancer patients
Revealing complex interactions among genetic lineages, DNA methylation clusters, and transcriptomic clusters in mGC
Given the strong heterogeneity of mGC, this study performed multi-regional sampling and single-cell multiomics sequencing to investigate the spatial distribution, genetic lineages, DNA methylation clusters, and transcriptomic clusters of cancer cells in mGC. Firstly, the genetic lineage of cancer cells within the same patient was accurately identified using the pattern of somatic copy number alterations. They found that DNA methylation clusters highly correlated with genetic lineages (Fig. 5). Within the same genetic lineage in a patient, different cancer cells exhibited similar DNA methylation patterns, while the DNA methylation levels varied significantly among cancer cells from different genetic lineages. This suggests that genome-wide demethylation is established during the formation of gastric cancer genetic lineage and can be maintained relatively stable during subsequent clonal expansion processes.
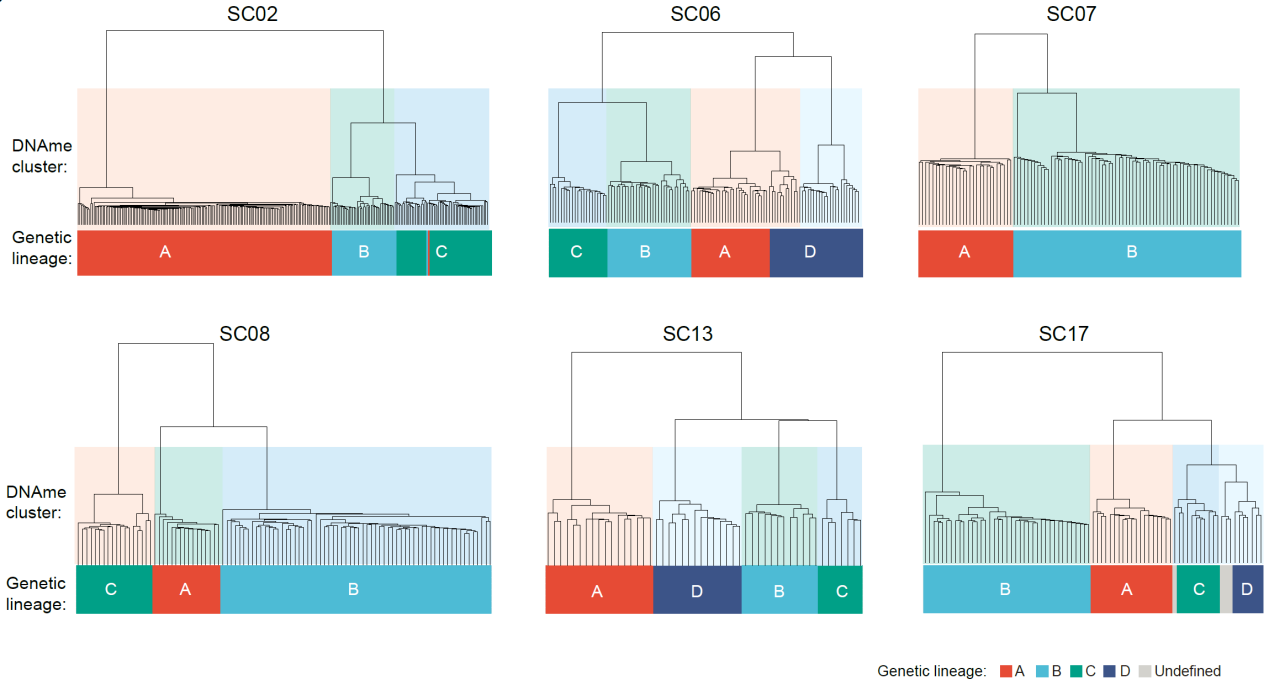
Fig. 5 Unsupervised hierarchical clustering of genome-wide DNA methylation profles generate cancer cell clusters highly consistent with their genetic lineages
However, within the same patient, the relationship between genetic lineages or DNA methylation clusters and transcriptomic subgroups is not a simple one-to-one correspondence. Cancer cells from the same genetic lineage can correspond to multiple transcriptomic clusters, and cancer cells from different genetic lineages can correspond to the same transcriptomic cluster. Furthermore, mGC exhibits strong spatial heterogeneity (Fig. 6). Not only can there be differences in genetic lineages, DNA methylation clusters, and transcriptomic clusters among cancer cells from different sampling regions within the same patient, but there can also be multiple distinct genetic lineages, DNA methylation clusters, and transcriptomic clusters within the same sampling regions.
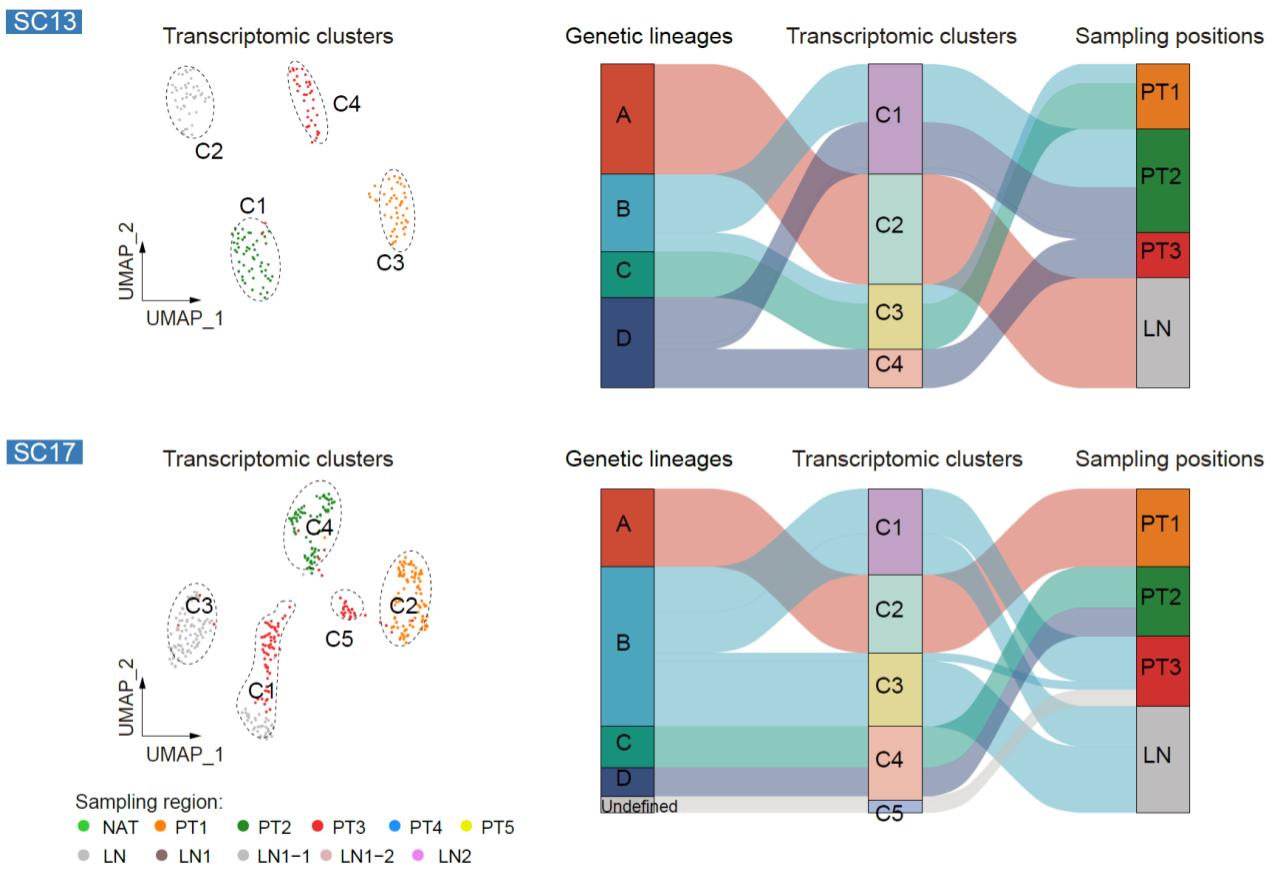
Fig. 6 The relationships among genetic lineages, DNA methylation clusters, and transcriptomic clusters
Key molecular features and immune microenvironment characteristics of cancer cells with different differentiation states in mGC
By analyzing the transcriptome data, the study identified the differentiation states of cancer cells in each transcriptomic cluster, including adenocarcinoma, neuroendocrine carcinoma, and hepatoid carcinoma, which were generally consistent with the pathological analysis results from the same sampling region. Furthermore, subdividing adenocarcinoma into different degrees of differentiation (well-differentiated, moderately differentiated, and poorly differentiated) posed a significant challenge due to several reasons. Firstly, due to the strong heterogeneity among gastric cancer patients, there is currently a lack of known and universal marker genes that can be used to identify different differentiation states of cancer cells within the same tumor. Secondly, despite the robust evidence provided by the pathological analysis results from each sampling region, spatial heterogeneity can still exist within the same sampling region. To address this issue, the study leveraged the advantages of multi-region sampling, point-to-point histopathological analysis, and single-cell multi-omics data analysis, and found the PCA's PC3 axis can separate the transcriptomic clusters of cancer cells at different differentiation states, including distinguishing different degrees of differentiation within adenocarcinoma (well-differentiated, moderately differentiated, and poorly differentiated). These results were generally consistent with the pathological analysis results from the same sampling region (Fig. 7).
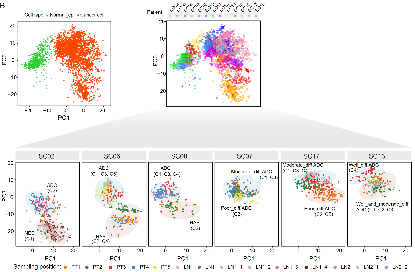
Fig. 7 The PCA projection using the transcriptome data from single-cell multiomics sequencing
Upon accurately identifying the differentiation states of cancer cells, the study further investigated the molecular features of cancer cells at different differentiation states (Fig. 8). The results revealed that the mucin family member MUC1 was highly expressed in moderately differentiated and well-differentiated adenocarcinoma, while its expression significantly decreased as the differentiation state worsened within each patient. Fibronectin (FN1) showed significantly increased expression in cancer cells with poorer differentiation states (neuroendocrine carcinoma and hepatoid carcinoma). Additionally, changes in chromatin accessibility and DNA methylation in gene promoter regions partially explained the differential expression levels of certain genes across different differentiation states.
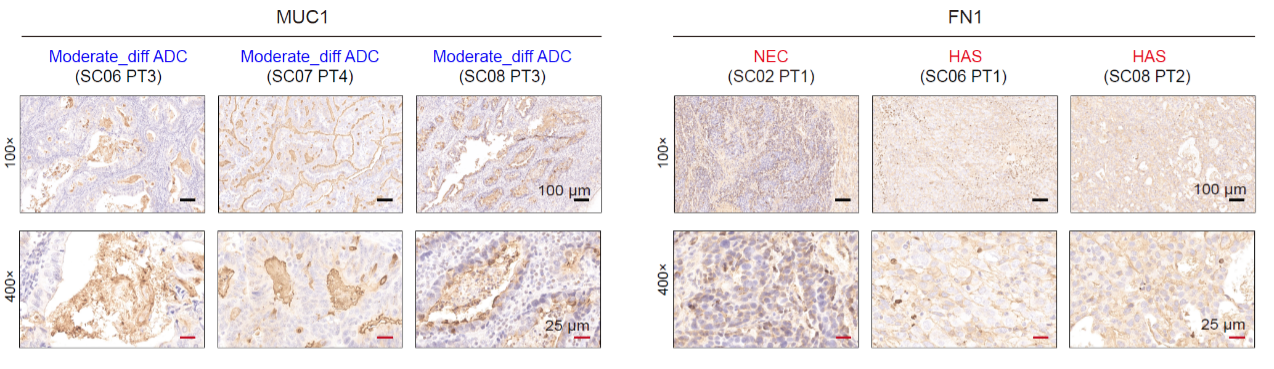
Fig. 8 The IHC staining of MUC1 and FN1 protein of some representative positions of mGC patients. NEC, neuroendocrine carcinoma; HAS, hepatoid carcinoma.
Furthermore, despite the strong inter-patient heterogeneity in the marker genes of differentiation states, they converged on some common signaling pathways. For example, genes upregulated in poorly differentiated cancer cells within each patient were enriched in pathways related to cell cycle and stress response, while genes upregulated in well-differentiated cancer cells within each patient were enriched in immune-related pathways.
The study found that in the primary tumor cells of most patients with mixed-type gastric cancer (5 out of 6), poorly differentiated cancer cells exhibited significantly higher global DNA methylation levels compared to well-differentiated cancer cells but still lower than those of normal gastric epithelial cells (Fig. 9). This suggests that the extensive global DNA demethylation occurring in the early stages of gastric cancer development leads to the aberrant transcription of repetitive sequences, represented by LINE-1, activating the antigen presentation of gastric cancer cells. This enables immune cells to recognize these cancer cells. However, during the process of further dedifferentiation or transdifferentiation into neuroendocrine carcinoma or hepatoid carcinoma, the global DNA methylation levels in these cancer cells partially rebound, potentially suppressing the aberrant transcription of repetitive sequences and reducing the antigen presentation characteristics of cancer cells. As a result, immune cells find it more difficult to recognize and attack these poorly differentiated or transdifferentiated cancer cells.
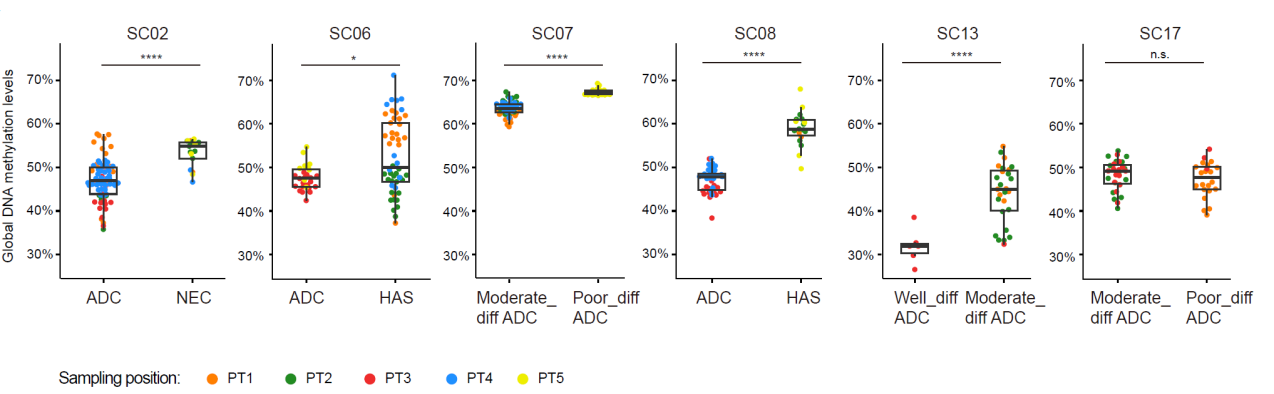
Fig. 9 The global DNA methylation levels of cancer cells with different differentiation states of six mGC patients
By comparing gastric cancers that simultaneously contain both adenocarcinoma and non-adenocarcinoma components, it was observed that the infiltration level of CD8+ T cells was lower in non-adenocarcinoma (neuroendocrine carcinoma and hepatoid carcinoma), with CD8+ T cells primarily located at the tumor margins, indicating an immune-excluded immune microenvironment (Fig. 10). Conversely, adenocarcinoma exhibited relatively higher levels of CD8+ T cell infiltration. Accordingly, the immune-related pathway scores in adenocarcinoma were significantly higher than those in non-adenocarcinoma (neuroendocrine carcinoma and hepatoid carcinoma).
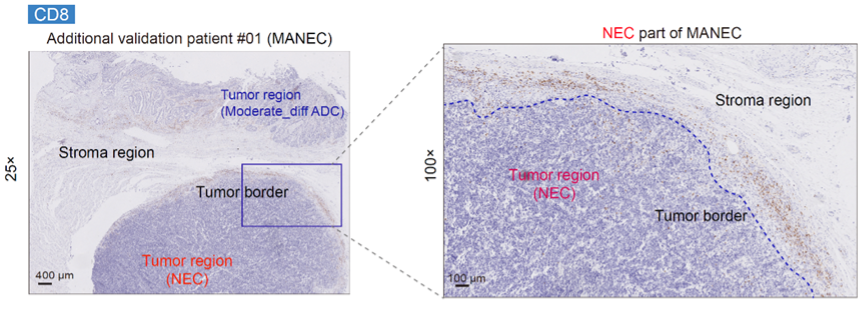
Fig. 10 IHC staining of CD8 protein for an additional validation patient with mixed adeno-neuroendocrine carcinoma
In conclusion, this study performed systematic multi-region sampling and single-cell multi-omics (genome, DNA methylation, chromatin accessibility, and transcriptome) sequencing analysis of matched samples including normal adjacent tissues, primary tumors, and lymph node metastases from gastric cancer patients who underwent radical surgery. It revealed common patterns of abnormal gene expression in cancer cells across different gastric cancer patients, indicating the widespread overexpression of intestinal epithelial cell-specific genes in different subtypes of gastric cancer. This reflects the increased plasticity of gastric cancer cells during the dedifferentiation process, where they lose their normal physiological functions. The study also generated a single-cell DNA methylation atlas of gastric cancer and found that gastric cancer, similar to colorectal cancer, undergoes extensive genome-wide DNA demethylation during tumorigenesis, accompanied by aberrant hypermethylation in a small number of gene promoter regions. Additionally, it was observed that within the same gastric cancer, different cancer cells from the same genetic lineage exhibit similar DNA methylation levels, while there can be substantial differences in DNA methylation levels between cancer cells from different genetic lineages. This suggests that genome-wide DNA demethylation is largely completed in the early stages of gastric cancer tumorigenesis and remains relatively stable during the expansion of each genetic clone thereafter.
The study also found that there is not a simple one-to-one correspondence between genetic lineages or DNA methylation clusters of cancer cells and transcriptional clusters. Multiple transcriptional clusters can correspond to the same genetic clone, and different genetic clone cancer cells can correspond to the same transcriptional subgroup. In mGC, it was observed that poorly differentiated cancer cells (such as neuroendocrine carcinoma and hepatoid carcinoma) exhibit partial reversion of global DNA methylation compared to well-differentiated cancer cells. These poorly differentiated cells upregulate genes related to the cell cycle, stress response, and downregulate genes related to immune pathways, resulting in lower levels of immune infiltration.
This research was supported by the National Natural Science Foundation of China, Biomedical Pioneering Innovation Center, and the Center for Life Sciences.
Link:https://doi.org/10.1093/nsr/nwad094