Cell Research | scNanoATAC-seq: a long-read single-cell ATAC sequencing method to detect chromatin accessibility and genetic variants simultaneously within an individual cell
On October 11, 2022, Prof. Fuchou Tang's lab at Peking University published a paper in Cell Research titled scNanoATAC-seq: a long-read single-cell ATAC sequencing method to detect chromatin accessibility and genetic variants simultaneously within an individual cell. scNanoATAC-seq is the first technology to detect single-cell chromatin accessibility based on the third-generation sequencing (TGS) (single-molecule sequencing) platform. scNanoATAC-seq integrates the advantages of long-read single-molecule sequencing platform and single-cell chromatin accessibility sequencing technology (scATAC-seq: Single cell Assay for Transposase Accessible Chromatin with high-throughput sequencing), enabling the simultaneous detection of open chromatin states and genetic variants within an individual cell.

The scNanoATAC-seq expanded the knowledge of chromatin accessibility sequencing analysis. The scATAC-seq technology based on the next-generation sequencing platform (NGS) enriches only short fragments of genomic DNA (mainly 80-300 bp) on open chromatin regions, which are used as signals for open chromatin. However, long fragments of genomic DNA are also generated during cleavage and tagmentation open chromatin regions using excess Tn5 transposase, and it is unclear whether these long fragments contain information on open chromatin states.
This study developed a single-cell ATAC-seq sequencing technology based on single-molecule sequencing platform (shown in Figure 1) and explored its application to biological issues. First, using five human cell lines and in vivo human peripheral blood mononuclear cells, this study demonstrated that scNanoATAC-seq technology could accurately cluster different cell types based on open chromatin states like NGS scATAC-seq and reveal key regulatory features of chromatin accessibility (shown in Figure 2).
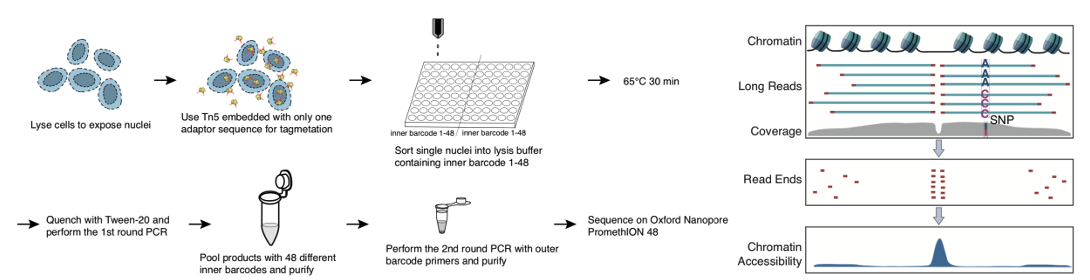
Figure 1 The workflow (left) and schematic diagram for peak calling (right) of scNanoATAC-seq.
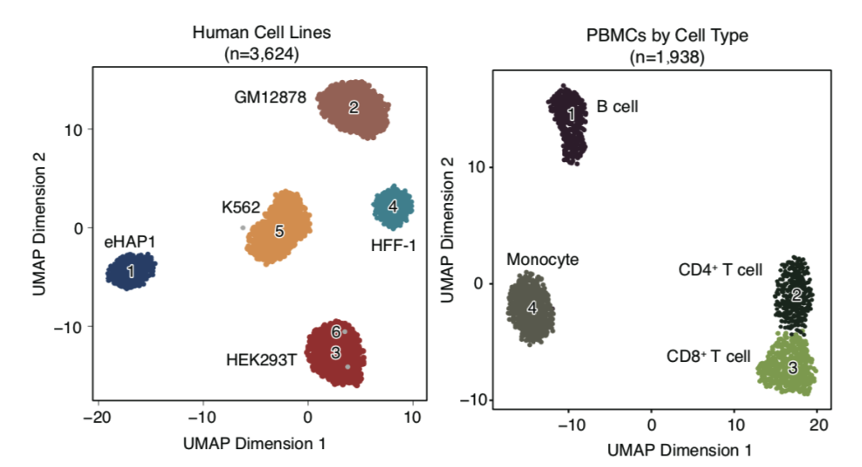
Figure 2 The results of scNanoATAC-seq technology to cluster different cell types
Next, the study took advantage of the long reads of scNanoATAC-seq technology to accurately identify allele-specific chromatin open regions. The vast majority of cell types in the human body are diploid cells. For diploid cells, the NGS ATAC-seq technology to distinguish two alleles in open chromatin regions requires heterozygous single nucleotide polymorphism (SNP) loci within that open chromatin region (between 80-300 bp in length). In contrast, the scNanoATAC-seq technology based on the third-generation sequencing platform to distinguish two alleles in open chromatin regions does not require heterozygous SNP sites within that open region, but only heterozygous SNP sites or heterozygous structural variants within 4000 bp on each side of that open region. In this way, the scNanoATAC-seq technology detected more than ten-fold allele-specific open chromatin regions in the same cell line compared to the NGS ATAC-seq technology. scNanoATAC-seq technology accurately genotyped chromatin accessibility signals (as shown in Figure 3) (the accuracy of detecting maternal alleles was 90.9%, and that of detecting paternal alleles was 88.1%). For example, applying the scNanoATAC-seq technique, this study detected an allele-specific open chromatin region on the promotor of imprinted differentially methylated gene TRIM61 in human B-lymphocyte line GM12878 (as shown in Figure 4, the chromatin in the promoter region of the maternal allele was open and the chromatin in the promoter region of the paternal allele was closed ), and this open chromatin region had no heterozygous SNP locus in the GM12878 cell line and couldn’t be detected by the short-read ATAC-seq method. Furthermore, this study found that the allele-specific open chromatin regions detected in GM12878 cell line using scNanoATAC-seq technique were mainly enriched on the X chromosome. This was consistent with previous findings using the DNase I hypersensitive loci assay. The reason for the tendency of allele-specific open chromatin occurred on the X chromosome was the much higher percentage of the cells with the paternal X chromosome silenced in the GM12878 cell line.
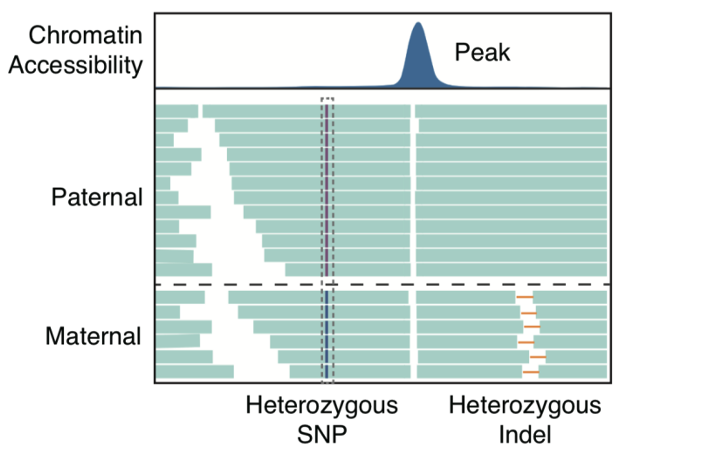
Figure 3 A schematic diagram of identifying allele-specific chromatin accessibility by scNanoATAC-seq.
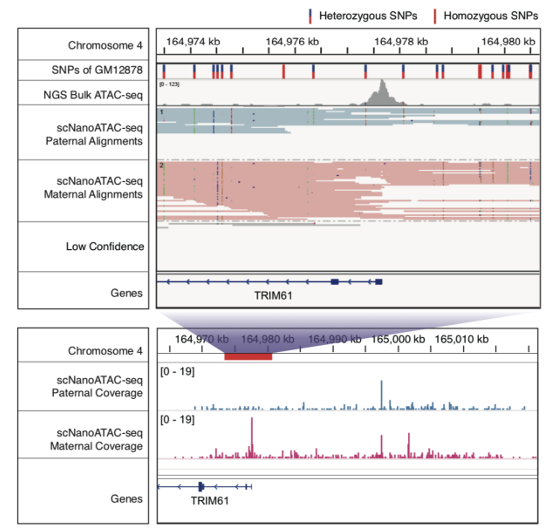
Figure 4 An allele-specific open chromatin peak on the promoter of TRIM61.
Subsequently, this study used scNanoATAC-seq technology to detect various structural variation events (insertions, deletions, duplications, inversions, translocations, etc.) in single cells. Using bulk ONT genome sequencing data of human chronic myeloid leukemia (CML) cell line K562 as a benchmark, scNanoATAC-seq technology detected 7,688 insertions (64.6% of the benchmark) and 6,120 deletions (67.7% of the benchmark) in the K562 cell line (supported by at least 5 single cells) with an accuracy of 93.8% and 75.5%, respectively. In addition to the classical BCR-ABL1 translocation event, the study could also detect an 89-kb deletion event that truncated both ZRANB1 and CTBP2 genes (as shown in Figure 5). CTBP2 was reported to inhibit leukemia proliferation, which implied that this study identified a potential loss-of-function SV of a tumor suppressor gene in K562 cells. In addition, scNanoATAC-seq technology also detected copy number variants in individual cells and accurately distinguished aneuploid cells from the normal diploid ones.
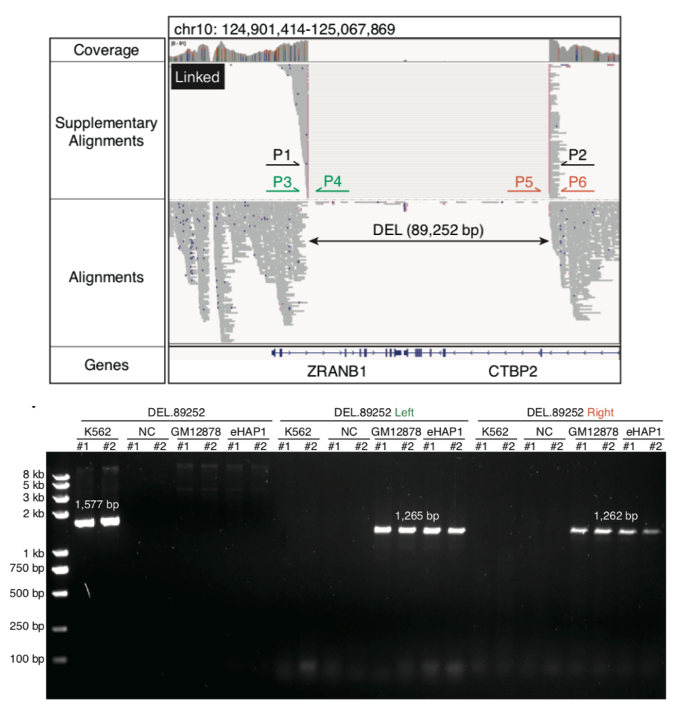
Figure 5 A somatic deletion truncating ZRANB1 and CTBP2 in K562 was detected by scNanoATAC-seq (top), which was validated as a homozygous somatic deletion by PCR products of the deletion region (P1 and P2), the left breakpoint (P3 and P4) and the right breakpoint (P5 and P6) (bottom).
Finally, this study exploited the long reads of scNanoATAC-seq to detect co-accessibility between pairs of neighboring peaks and 3,868 pairs of co-accessible peaks were detected in GM12878 cell line. Taking the co-accessible peak pair near the SOX4 gene locus as an example, these adjacent co-accessible peaks were supported by long reads directly connecting the two open regions, and the length distribution of peak-supporting reads altered compared to the background (as shown in Figure 6 and Figure 7). In contrast, the analysis of chromatin co-accessible events using NGS scATAC-seq technology is mainly dependent on the detection coverage (or detection sensitivity) of each single cell. If the detection coverage of scATAC-seq is relatively low, it will be difficult to detect chromatin co-accessible events. More importantly, even with high detection coverage of scATAC-seq, half of the chromatin co-accessible events are signaled by the co-accessible of two different alleles in a single cell (e.g., the enhancer of allele #1 "co-accessible" with the promoter of allele #2), The signals of chromatin co-accessible events detected by the scNanoATAC-seq technology were direct linkage information from a single DNA molecule and were real co-accessible events from the same allele in a single cell (e.g., enhancer of allele #1 co-accessible with the promoter of allele #1, or enhancer of allele #2 co-accessible with the promoter of allele #2), there were no such technical artifacts as described above.
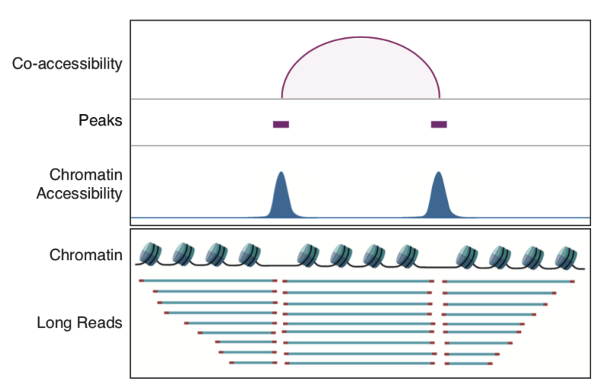
Figure 6 A schematic diagram of identifying co-accessibility peaks.
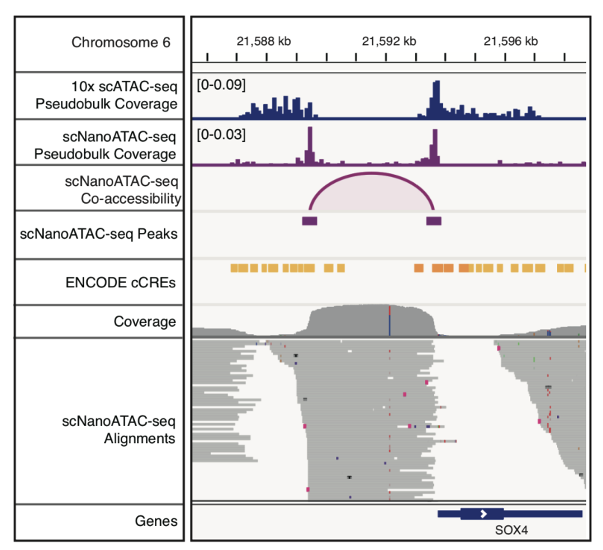
Figure 7 The co-accessible events near SOX4.
In summary, the scNanoATAC-seq technique has a wide range of biological applications. The method allows simultaneous detection of chromatin accessibility and genetic variations in a single cell. The method takes advantage of long reads to identify allele-specific open chromatin peaks without heterozygous SNP inside a peak, which is not feasible for short-read scATAC-seq. The method can also provide the direct evidence of co-accessibility between neighboring peaks from scNanoATAC-seq, where the chromatin accessibility of two sites in the same single cell and in fact on the same allele is detected simultaneously by a long read. scNanoATAC-seq technology heralds the era of single-molecular sequencing of single-cell epigenomics.
The higher the concentration of Tn5 transposase used in the DNA-exposed regions, the shorter the DNA fragments obtained. It is worth noting that when using excess Tn5 transposase, long fragments were also captured by the scNanoATAC-seq. It can be inferred that in addition to the usual open chromatin regions, these long fragments may capture permissive chromatin regions, which cannot be detected by short-read sequencing. In fact, more nucleosome occupancy pits were detected by scNanoATAC-seq compared with NGS ATAC-seq (as shown in Figure 8). In addition, scNanoATAC-seq may have better performance than NGS scATAC-seq on mapping sequencing reads to genomic repetitive elements. All of the above issues deserve to be explored in depth by applying the scNanoATAC-seq technology, which also reflects the necessity to develop the scNanoATAC-seq technology.
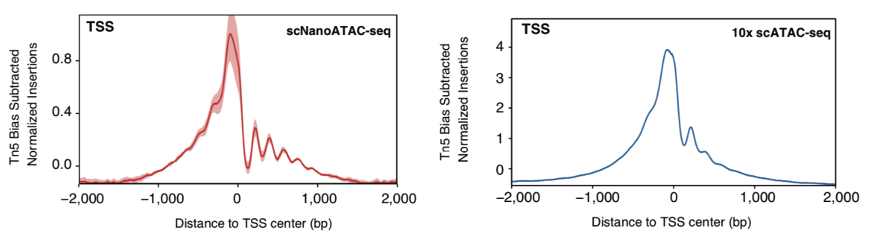
Figure 8 Signals of scNanoATAC-seq (left, red curves) and 10x scATAC-seq (right, blue curves) enriched around TSS.
Postdoctor Yuqiong Hu, Ph.D. candidates Zhenhuan Jiang and Kexuan Chen from the School of Life Sciences at Peking University contributed equally to the paper. Prof. Fuchou Tang from the Biomedical Pioneering Innovation Center of Peking University is the corresponding author of the paper. The research project was supported by the Beijing Advanced Innovation Center for Genomics and grants from National Key R&D Program of China.