Cancer Research | Single-cell dissection of the multiomic landscape of high-grade serous ovarian cancer
On August 15th, 2022, Tang Lab from Biomedical Pioneering Innovation Center (BIOPIC), Beijing Advanced Innovation Center for Genomics (ICG) collaborating with Heng Cui’s team from Peking University People’s Hospital and Kehkooi Kee’s team from Tsinghua University, published a paper titled Single-cell dissection of the multiomic landscape of high-grade serous ovarian cancer on Cancer Research , which systematically analyzed multiomics molecular characteristics and their mutual regulatory relationships of high-grade serous ovarian cancer.

Ovarian cancer is one of the leading causes of gynecological cancer death. High-grade serous cancer (HGSC) is the most aggressive and common subtype of ovarian cancer (comprising approximately 70% of all cases). HGSC is generally diagnosed at an advanced stage, presenting with extensive metastases and massive ascites. Despite an initial clinical response to the surgery and chemotherapy, most patients will relapse within 5 years. Hence, understanding the pathological mechanisms of HGSC initiation, promotion, and progression will be of great value. Intratumor heterogeneity is one of the most challenging obstacles to study HGSC. Single-cell sequencing technique provides a powerful tool to study intratumoral heterogeneity. Previous studies have revealed the strong intratumor heterogeneity of HGSC, while the alterations of genome and epigenome during HGSC promotion and progression remain to be comprehensively analyzed in HGSC.
Several key conclusions emerge from this work:
The activities of oxidative phosphorylation pathway and glycolysis pathway are enhanced simultaneously during HGSC promotion.
Through differential expression analysis between fallopian tube epithelial (FTE) cells and primary tumor cells, the research identified that 361 genes were upregulated in primary tumors, which were mainly enriched in pathways associated with VEGFA-VEGFR2 signaling, metal ion homeostasis, regulation of viral process, and metabolism regulation. Of note, genes associated with both of oxidative phosphorylation (OXPHOS) and aerobic glycolysis were both upregulated. The research calculated OXPHOS and glycolysis scores for each cell (Figure 1), and identified that cancer cells could not be further separated into subtypes, suggesting that cancer cells simultaneously elevated the expression of both OXPHOS and aerobic glycolytic genes compared with FTE cells.
To further clarify the typical characteristics of HGSC, the research also compared HGSC with non-HGSC tumors. The HGSC tumors could be clearly separated from non-HGSC tumors by principal component analysis (PCA), demonstrating that HGSC had very distinct transcriptional patterns. Furthermore, the differential expression analysis showed that expression of metallothionein genes and interferon (IFN) signaling pathway genes were also increased in HGSC when compared with non-HGSC tumors (Figure 1), indicating that the strong upregulation of interferon signaling pathway is a prominent characteristic of high-grade serous ovarian cancer.
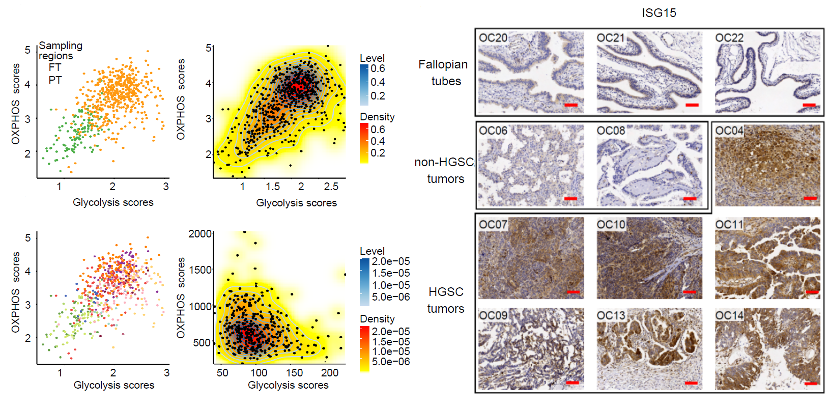
Figure 1. The OXPHOS and glycolysis scores of single cells and immunohistochemical staining of ISG15.
The amplification of chromosome 8 leads to increased dose of MYC gene during tumorigenesis of high-grade serous ovarian cancer, which further contributed to the enhanced activity of oxidative phosphorylation pathway and glycolysis pathway.
HGSC frequently carried strong amplification on chromosome 8 (Chr8). The research identified that the chromosome 8 was the most enriched chromosome of genes which were upregulated in primary tumor cells compared with FTE cells. In addition, the research observed that MYC (located on Chr8), which regulated cell division, OXPHOS and aerobic glycolysis, was upregulated in 10 out of the 12 HGSC patients. The results suggested that the amplification of chromosome 8 may contribute to tumorigenesis of HGSC through amplifying the master regulator MYC and increasing the dosages of these 46 genes related to OXPHOS.
The regulation of DNA methylation on gene expression.
DNA methylation is one of the most important epigenomic modifications, which undergoes extensive reprogramming during tumorigenesis and may impact gene expression. The research analyzed the potentially regulatory effects of repeat element methylation, promoter methylation, and distal region methylation on gene expression.
The results showed that most patients (9/11) have lower global DNA methylation levels in ovarian cancer cells than those in normal FTE cells. For all patients, both satellites and long interspersed nuclear element 1 (LINE1) exhibited the most dramatic DNA demethylation. Correlation analysis showed that genes that may be negatively regulated by both satellites and LINE1 were mainly enriched in metallothionein bind metals, interferon, apoptosis and angiogenesis related pathways; the genes that might be positively regulated were mainly enriched in the pathways related to ribosome synthesis.
Further analysis of genes potentially regulated by promoter methylation revealed that genes related to metallothionein binds metal and interferon were also affected by promoter methylation.
The research used MICMIC to identify methylation regulation networks of distal regulatory regions. We detected a total of 16,941 regulatory region-target pairs. Of these pairs, 53.9% were anticorrelated with the expression of target genes. Most target genes were potentially regulated by DNA methylation of less than 5 regulatory regions. For example, the upregulated gene IFI27 (Figure 2), whose expression was negatively correlated with its promoter DNA methylation, was also negatively correlated with methylation of a distal region. In addition, this distal region is overlapped with a known ovarian enhancer and its flanking regions exhibit higher chromatin accessibility in primary tumor cells compared with FTE cells.
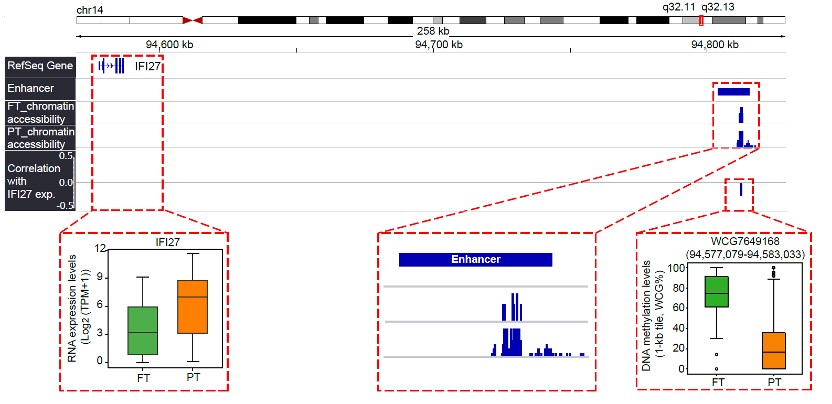
Figure 2. Genome browser view showing that a distal region (WCG7649168: chr14: 94,577,079-94,583,033) potentially regulated the expression of IFI27.
The regulatory activities of FOXK1, TFAP2C, NR2F6, DDIT3 and other important transcription factors are significantly enhanced during the tumorigenesis of high-grade serous ovarian cancer.
To explore potential key regulators in cancer cells, the research performed TF binding motif enrichment of NDRs using chromVAR (22). In total, motifs of 107 TFs were more open in cancer cells, while 126 were more closed (Figure 3). Of these 107 TFs, expression levels of 4 TFs in cancer cells are significantly higher than that in FTE cells: FOXK1, TFAP2C, NR2F6, and DDIT3. FOXK1 is an important regulator to induce aerobic glycolysis. Considering that the glycolysis pathway was upregulated in HGSC and the binding motifs of its transcriptional regulator FOXK1 exhibited high accessibility, we suggested that FOXK1 could be an attractive candidate therapeutic target for HGSC.
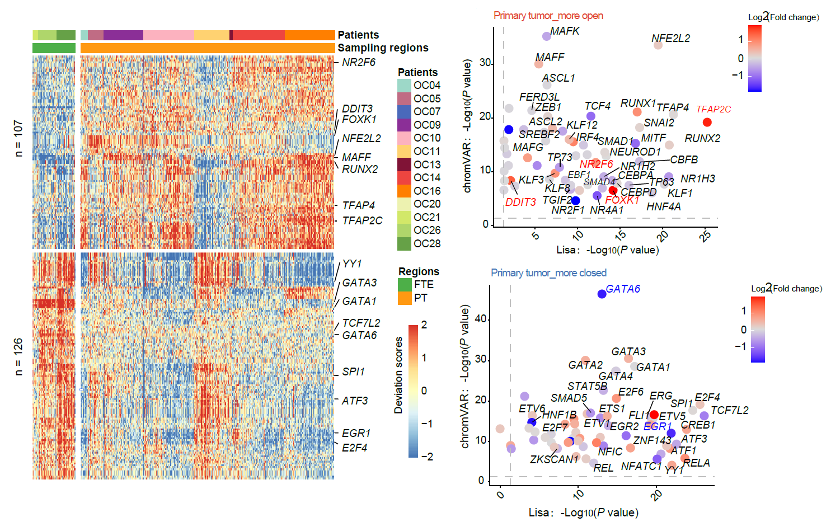
Figure 3. Integrated analysis using data of chromatin accessibility and transcriptome to identify the important regulatory transcription factors in HGSC.
The gene expression profiles of peritoneal metastases are similar to their matched primary tumors.
Results showed that very few genes were differentially expressed for most patients (5/6). Only a patient with lymphatic node metastases had more than 2000 differentially expressed genes. These results proposed that the microenvironment of peritoneal metastases did not strongly affect the global RNA expression patterns of cancer cells, though the cancer cells had invaded and proliferated in a different organ, and implied that the metastatic capacity was probably acquired at an early stage during tumorigenesis.
Though only a few DEGs between metastasis and primary tumors were shared by different patients, these genes might provide some clues to identify critical genes promoting or suppressing metastasis. This study demonstrated the role of downregulation of HSPA6 in metastases of high-grade serous ovarian cancer (Figure 4). The results showed that overexpression of HSPA6 could inhibit the migration and invasion of cancer cells, suggesting that downregulation of HSPA6 promoted the tumor metastasis process of high-grade serous ovarian cancer.
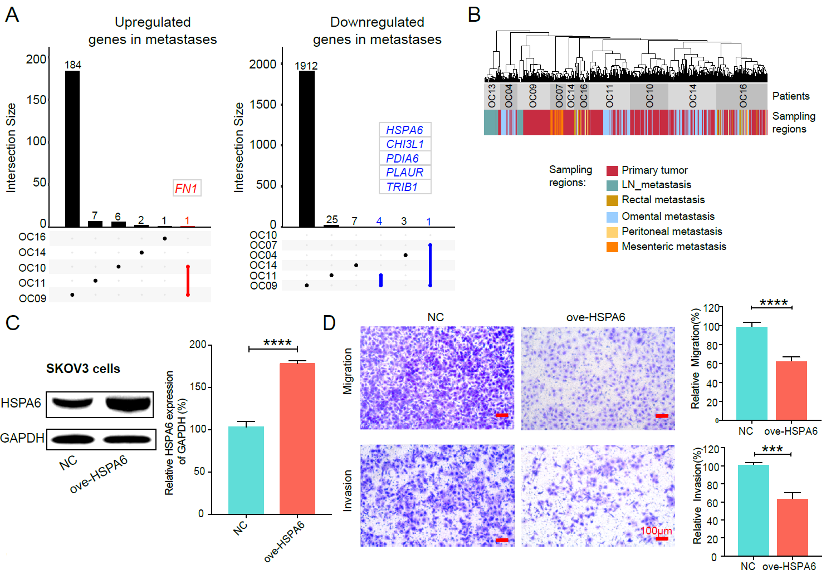
Figure 4. The differentially expressed genes between primary tumors and matched metastases as well as the functional validation of HSPA6.
Cancer cells maintained DNA methylation levels and patterns during peritoneal metastasis.
The research identified that the global DNA methylation levels of cancer cells were stable during metastasis in most patients (Figure 5). Thus, they speculated that cancer cells maintained the global methylation characteristics of their genetic lineages during metastasis. The unsupervised hierarchical clustering of DNA methylation confirmed that DNA methylation patterns produce similar lineage histories to those inferred by SCNAs.
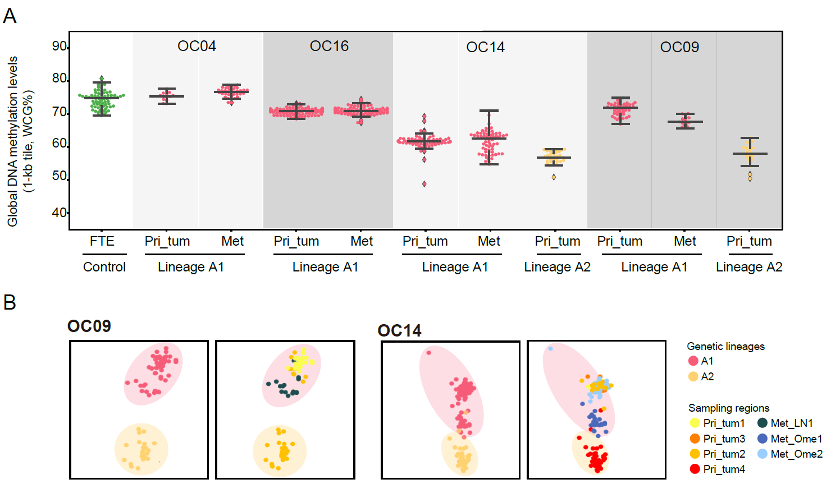
Figure 5. Cancer cells maintained DNA methylation levels and patterns during metastasis.
The gene expression differences between tumor cells of different lineages within the primary tumor can be used to search for candidate genes involved in tumor metastasis.
Different lineages not only have different SCNAs, DNA methylation levels and RNA expression patterns, but may also have different metastatic potential. For example, both patient OC09 and OC14 harbored two genetic lineages (lineage A1 and A2) of cancer cells, and the lineage A1 had established secondary tumors, but the other lineage (lineage A2) had not. Differential expression analysis revealed that CCN1 and HSP90AA1 were upregulated in the metastasized lineage A1 for both OC09 and OC14. Results showed that knockout of CCN1 significantly restrained the migration and invasion of cancer cells by wound healing assay and transwell assays (Figure 6). Inhibition of HSP90AA1 using the HSP90α revealed that TAS-116 could inhibit ovarian cancer cell migration and invasion (Figure 6). Together, the results demonstrated that CCN1 and HSP90AA1 may participate in the metastasis of HGSC and may serve as potential therapeutic targets for ovarian cancer.

Figure 6. Functional validation of CCN1 and HSP90AA1.
In summary, this study provided the first systematic exploration about inter- and intratumor heterogeneities of SCNAs, DNA methylome, chromatin accessibility, and the transcriptome of primary tumors and matched metastases in high-grade serous ovarian cancer at single-cell and single-base resolution. They integratedly analyzed the multiomic changes of high-grade serous ovarian cancer, and identified the candidate targets (such as CCN1 and HSP90AA1), which provides insights into the molecular characteristics of this disease, and could help improve diagnosis and treatment.
Ph.D. candidate Yicheng Wang and Ph.D. candidate Haoling Xie from Biomedical Pioneering Innovation Center, School of Life Sciences of Peking University, Prof. Xiaohong Chang and Prof. Yi Li from Peking University People’s Hospital, as well as Ph.D. Wenqi Hu and Ph.D. candidate Mengyao Li from Tsinghua University are the co-first authors of the paper. Professor Fuchou Tang from Biomedical Pioneering Innovation Center, School of Life Sciences of Peking University, Professor Heng Cui from Peking University People’s Hospital, and Professor Kehkooi Kee from Tsinghua University are corresponding authors. This research project was supported by the National Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics.