Nucleic Acids Research | De novo assembly of human genome at single-cell levels
On July 12th, 2022, Tang Lab from Biomedical Pioneering Innovation Center (BIOPIC) and Beijing Advanced Innovation Center for Genomics (ICG) published a paper titled “De novo assembly of human genome at single-cell levels” on Nucleic Acids Research , which completed the human genome assembly with high continuity at single-cell levels, and explored the impact of different assemblers and sequencing strategies on genome assembly.

Usually, long-read sequencing assembly requires large amounts of DNA (typically several micrograms from millions of cells), and therefore most human genome assemblies have been restricted to bulk genome sequencing datasets without keeping the potential genetic heterogeneities among individual cells. However, this is impractical under many situations. In practice, at least two basic challenges need to be addressed.
The first is about cell heterogeneity. Bulk data assembly is based on the premise that all cells in a bulk sample carry the same genome; otherwise, it would be difficult to discriminate variations among different genetic clones and variations among different haplotypes within a cell.
The second challenge is that in most cases only a small number of genomic DNAs can be obtained for sequencing, such as in early embryonic development studies and cancer genome research. These cells are difficult to culture and amplify in vitro ; even if they could be cultured, there is no guarantee that their genome structure during in vitro will remain the same as in vivo .
Assembling the human genome using a small number of genomic DNAs or even a single cell genome sequencing data is much more challenging. It requires not only the support of single-cell TGS platform-based genome sequencing technology, but also a good data analysis strategy and a suitable assembler.
The authors employed SMOOTH-seq on PacBio HiFi and Oxford Nanopore Technologies (ONT) platforms to sequence K562 (a human chronic myelogenous leukemia cell line) and HG002 (a normal diploid lymphoblast cell line) and demonstrate the feasibility of genome assembly based on scWGS dataset with different assemblers and rigorous evaluations.
Several key conclusions emerged from this work:
1. The SMOOTH-seq was comprehensively optimized to be suitable for both PacBio and ONT single-molecule sequencing platforms. Previously, SMOOTH-seq was only applied on PacBio HiFi platform, and the optimized SMOOTH-seq can also be used on ONT platform, which expanded the application of SMOOTH-seq and balanced the accuracy and the cost of sequencing.
2. Using recently emerged assemblers like hifiasm, Hicanu and wtdbg2 to assemble the human K562 cell line genome from 95 individual cells sequencing data (PacBio HiFi platform). For primary contigs, Hifiasm assembly has the longest NG50 (the sequence length of the shortest contig at half of the reference genome size) with 2.11Mb, the highest QV (a log-scaled probability of error for assembly) value of 42.5, the highest BUSCO completeness (94.7%), and the longest contig from Wtdbg2 can reach 14.12Mb. In addition, assembly contig can span most of the major histocompatibility complex (MHC) region (Figure 1).
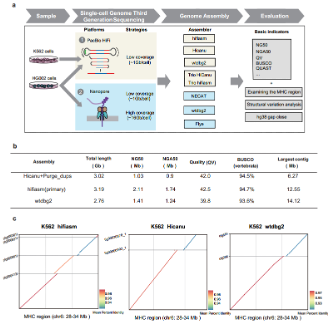
Figure 1. The assembly workflow and K562 assembly metrics.
3. Using hifiasm, Hicanu and wtdbg2 to assemble the human HG002 cell line genome from 157 individual cells sequencing data (PacBio HiFi platform). For primary contigs, Wtdbg2 has the highest continuity with NG50 of 0.65 Mb, the largest contig of 6.82 Mb and the highest BUSCO completeness (91.1%). Then, the authors used trio HiCanu and trio hifiasm to conduct the trio-based haplotype assembly. Through the assembly of wtdbg2, the NG50 size of the trio HiCanu F1 haplotigs was 0.28 Mb (HG003 haplotype) and 0.31 Mb (HG004 haplotype), and both parental haplotypes’ BUSCO completeness was greater than 84%, and QV value >36. In addition, for the six classical human leukocyte antigen (HLA) genes assembly, Trio HiCanu can capture most of them correctly (Figure 2).
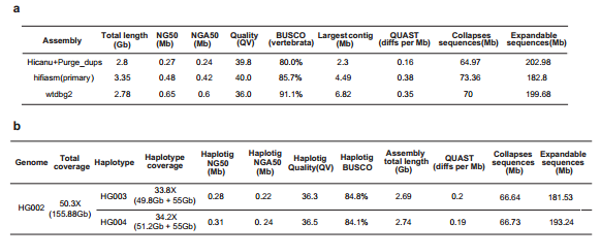
Figure 2. Single-cell haplotype assembly metrics of HG002 cells (PacBio HiFi platform).
4. Using Flye,Necat,wtdbg2 to assemble the human HG002 cell line genome from 192 individual cells sequencing data (ONT platform). Flye shows its fitness in single-cell datasets, which has the highest continuity with NG50 of 1.38 Mb, and its BUSCO completeness was 93.1% and the largest contig is 11.42 Mb. According to the results, for the assembly of single-cell ONT dataset, the choice of assembler can drastically affect the quality of the final results. Furthermore, the assembled contigs from Flye closed 39 gaps, where the length of 14 gaps is greater than or equal to 50 kb according to the annotation of hg38 (Figure 3).
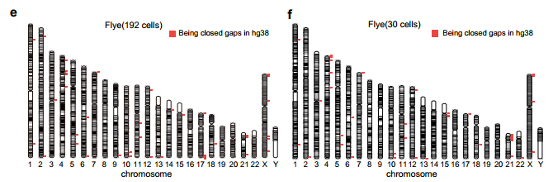
Figure 3. Assembly metrics for HG002 192 cells at a lower sequencing depth per cell.
5. Using Flye, wtdbg2 to assemble the human HG002 cell line genome from 30 individual cells sequencing data (ONT platform; high coverage). To investigate the lower limit of numbers of single cells that need to be sequenced for genome assembly, the authors deeply sequenced 30 HG002 single cells (average sequencing depth of a single cell was 5×) with high genome coverage (ranging from 27.9% to 53.4%) on ONT platform. They then explored the results of assembly of 30 cells, 20 cells, 10 cells and even just a single cell. The results showed that the assembly continuity with sequencing data from 30 individual cells (average genome coverage ∼ 41.7%) can achieve the NG50 of 1.35 Mb, the longest contig was ∼11 Mb and BUSCO completeness was ∼92%. Furthermore, the assembled contigs from Flye closed 38 gaps, of which 15 gaps’ lengths are greater than or equal to 50 kb (Figure 4).
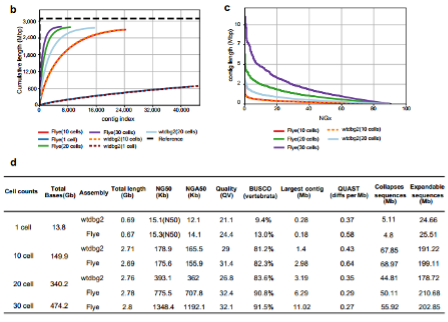
Figure 4. Assembly metrics for HG002 30 cells at a higher genome coverage.
6. Through analyzing the structural variations (SVs) of the assembled genome of K562 cells, many more insertion events could be identified and complex structural variations could be more efficiently and accurately illustrated.
The authors used both primary and alternate contig sets from hifiasm and Hicanu to detect SVs, and finally identified 7132 insertions and 6365 deletions from hifiasm, 6604 insertions and 4355 deletions from Hicanu. The precision and true positive rate of insertion and detection for these two assemblers were both >0.9 and >0.7 respectively. Moreover, translocation events could be more reliably identified after genome assembly. For example, translocation of chr3 with chr10 generates CDC25A-GRID1 fusion gene, which is not detected in single cell direct mapping results, but after assembly, a contig of 3.7 Mb permitting us to find this event robustly (Figure 5). To further verify the results, the authors selected 20 SVs (6 deletion events and 14 insertion events) detected from both HiCanu and hifiasm but not detected at the single cell direct mapping data as candidates for SV validation, and the verification accuracy is up to 80% (Figure 6).
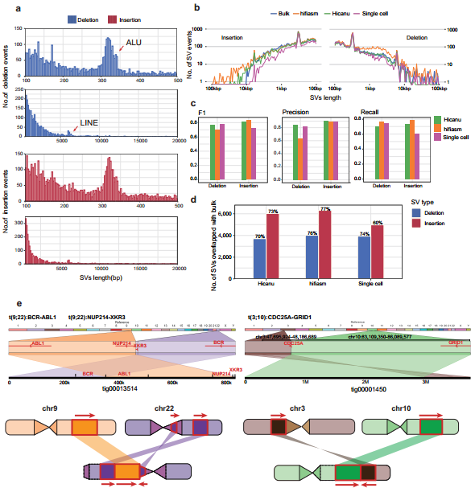
Figure 5. SVs discovery and distribution from K562 cells assembly.
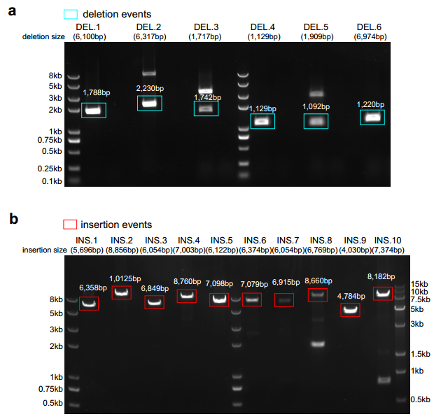
Figure 6. PCR validation of 16 SV events.
In summary, the availability of few cells and the large genetic heterogeneity within a population of cells are two difficulties that hinder the application of genome assembly in biomedical research. Single cell whole genome long-read sequencing technology can help us solve these problems. The authors have used different sequencing platforms and strategies to explore the feasibility of single-cell de novo genome assembly and identified the factors affecting the assembly results, and finally improved the resolution of genome assembly to single-cell levels. With further development of single-cell whole genome long-read sequencing technologies, restoring genome structure from just a single cell will finally be achieved, and will promote related biomedical research.
Haoling Xie (Ph.D. candidate) from the School of Life Sciences of Peking University and Wen Li (Ph.D. candidate) from the Academy for Advanced Interdisciplinary Studies of Peking University are the co-first authors of this paper. Professor Fuchou Tang of Beijing Advanced Innovation Center for Genomics, Biomedical Pioneering Innovation Center is the corresponding author. This research project was supported by the National Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics.