Cell Discovery ∣ Integrated single cell multiomics analysis reveals novel candidate markers for prognosis in human pancreatic ductal adenocarcinoma
On Feb. 15th, 2022, Tang Lab from Biomedical Pioneering Innovation Center (BIOPIC) and Beijing Advanced Innovation Center for Genomics (ICG) published a paper titled “Integrated single cell multiomics analysis reveals novel candidate markers for prognosis in human pancreatic ductal adenocarcinoma” on Cell Discovery, which systematically revealed the critical epigenomic features of cancer cells in PDAC patients at the single-cell level.

Pancreatic cancer, one of the most lethal type of cancers, is also one of the most difficult cancers to detect. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, and surgical resection is the only chance for cure; however, no more than 20% of PDAC patients are eligible for this treatment strategy. Although previous studies based on genome-wide analysis and gene expression profiles revealed complex molecular mechanism in pancreatic cancer, these data were quite limited for comprehensive analysis, especially for identifying the global features for PDAC cancer cells. The epigenetic characteristics of PDAC cancer cells remain largely elusive due to their extremely high intratumoral stromal content.
Several key conclusions emerge from this work:
1. Normal epithelial cells provided an accurate normal reference for studying the characteristics of cancer cells.
We used an improved version of our single-cell multiomics sequencing method scCOOL-seq that can simultaneously assess the genome (copy number variations), DNA methylome, chromatin accessibility, and transcriptome in the same individual cell. Different from previous studies that only determine normal cells by RNA expressions, we used the multiomics data to precisely define the non-cancer epithelial cells. In this way, we obtained significant proportions of cells as “normal epithelial cells” inside the PDAC tumor lesions exhibiting normal diploid genome, and their DNA methylation and chromatin accessibility features were compared with those of normal epithelial cells in the tumor adjacent tissues (Fig. 1). These normal epithelial cells were naturally presented in tumor tissue. They can be used as an accurate reference control for studying molecular characteristics of tumor cells, which solves the difficulty of obtaining matched control samples for PDAC. In addition, our results showed that the DNA methylation levels of cancer cells in PDAC were generally lower than normal epithelial cells, with a reduction of 5% to 20%, which was different from that of colorectal cancer (7% to 36%).
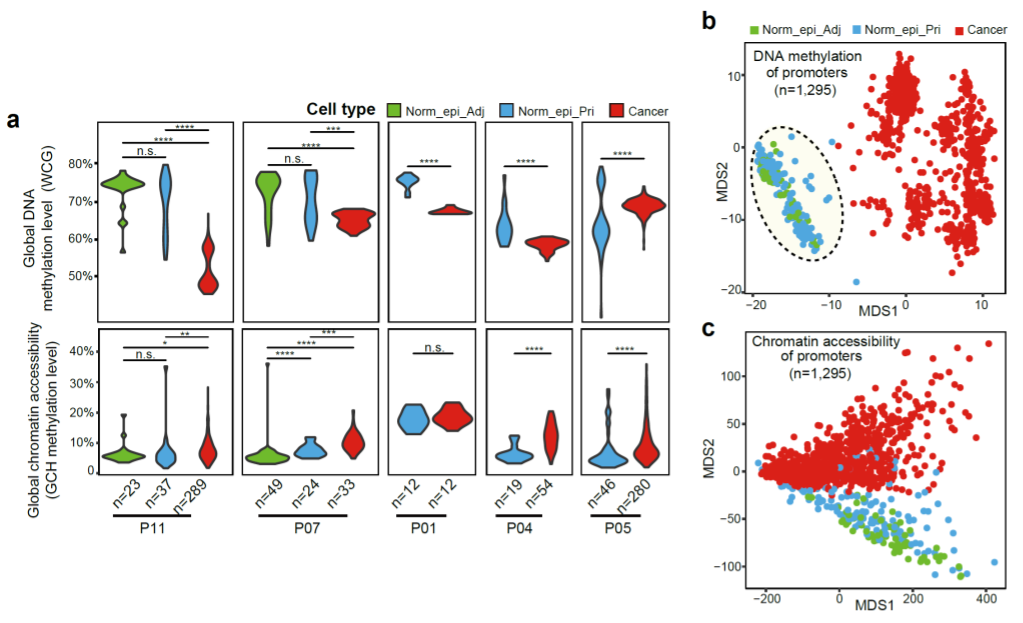
Fig 1. The global DNA methylation level and chromatin accessibility in the primary PDAC tissues.
2. The DNA methylation changes of PDAC.
All 13 patients showed more genes with increased promoter DNA methylation than genes with decreased promoter DNA methylation in cancer cells (Fig. 2). Many of the hypermethylated gene promoters were shared across different patients, and they were enriched for genes relating to the neural system by Gene Ontology analysis. A subset of 77 differentially methylated promoters showed clear negative correlation between promoter DNA methylation and gene expression in the cancer cells we have analyzed. These results suggest that using DNA methylation inhibitors reactivates these genes which are commonly silenced by DNA methylation in different patients.
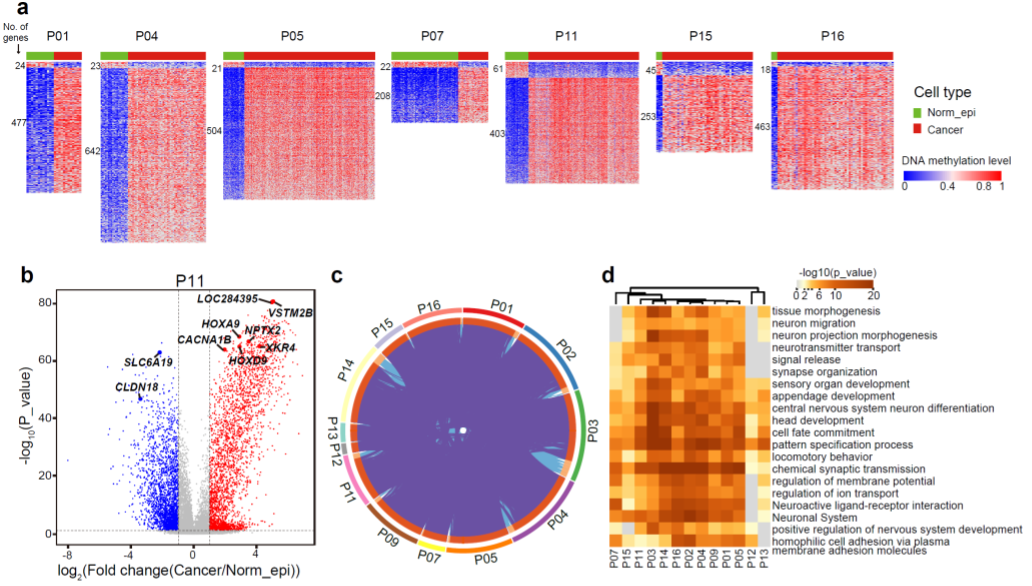
Fig. 2 Promoter regions in cancer cells show global DNA hypermethylation.
3. Relationship between DNA methylation and RNA expression in PDAC.
When analyzing the cross-omics relations, we found that the positive correlation between RNA expression and corresponding gene body DNA methylation was clearly stronger in cancer cells than that in normal epithelial cell (Fig. 3). We also found that the negative correlations between RNA expression and promoter DNA methylation of corresponding genes were much stronger in cancer cells than in Norm_epi cells. These results indicate that DNA methylation enhances the regulation of gene expression in tumorigenesis of PDAC.
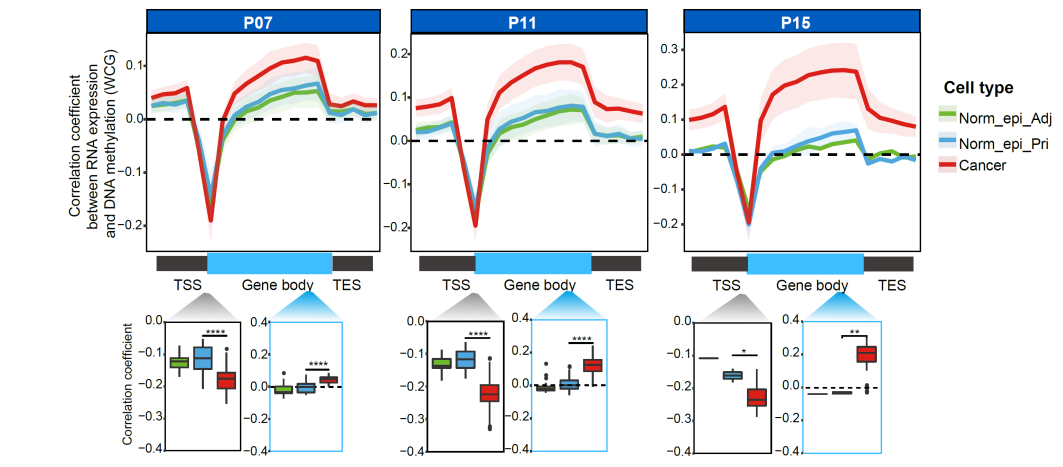
Fig. 3 DNA methylation shows a stronger correlation with RNA expression in cancer cells.
4. Global DNA demethylation enriched in heterochromatin regions in cancer cells
The DNA demethylation levels in cancer cells were positively correlated with the densities of LINE-1 but negatively correlated with densities of H3K4me3, H3K27Ac and H3K36me3 (Fig. 4), indicating that the DNA demethylation in PDAC cells was strongly enriched in the heterochromatin regions (LINE-1-enriched regions), but depleted in the euchromatin regions (H3K4me3-, H3K27Ac-, or H3K36me3-marked regions). These results suggested that tumorigenesis of PDAC might involve the heterochromatin disorganization, including their prevalent DNA demethylation, similar to the situation in colorectal cancer, as we previously showed.
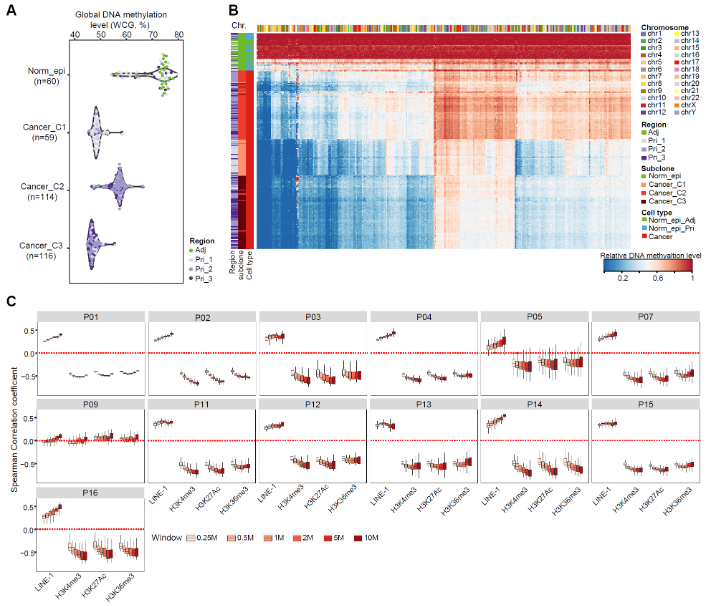
Fig. 4 Global DNA demethylation enriched in heterochromatin regions in cancer cells.
5. Combined multiomics analysis revealed that key transcription factors play an important role in tumorigenesis of PDAC.
We found the extent of open chromatin states in promoter regions was positively correlated with the RNA expression levels of the corresponding genes in all cell types we have analyzed (Fig. 5). We identified 64,339 and 265,213 nucleosome-depleted regions (NDRs, the genomic regions with local open chromatin features) in Norm_epi cells and cancer cells, respectively. Majority of the cancer specific NDRs were patient-specific and only a small percentage of the cancer specific NDRs were shared between different patients. We further searched for significantly enriched TF binding motifs using cancer-specific and Norm_epi-specific NDRs separately and identified candidate regulatory TFs potentially promoting or suppressing PDAC tumors. FOSL2, with higher RNA expression in cancer cells, also showed to be significantly correlated with poorer overall survival of the patients. Notably, the neural developmental related TFs, which showed consistent promoter hypermethylation in cancer cells of all patients enriched binding motifs in the normal cells as well, such as ASCL1, CUX1/2, DLX1/2/3/5, NEUROD1, NKX6-1, NEUROG2, suggesting that dysregulation of these transcription factors may be involved in tumorigenesis of PDAC.
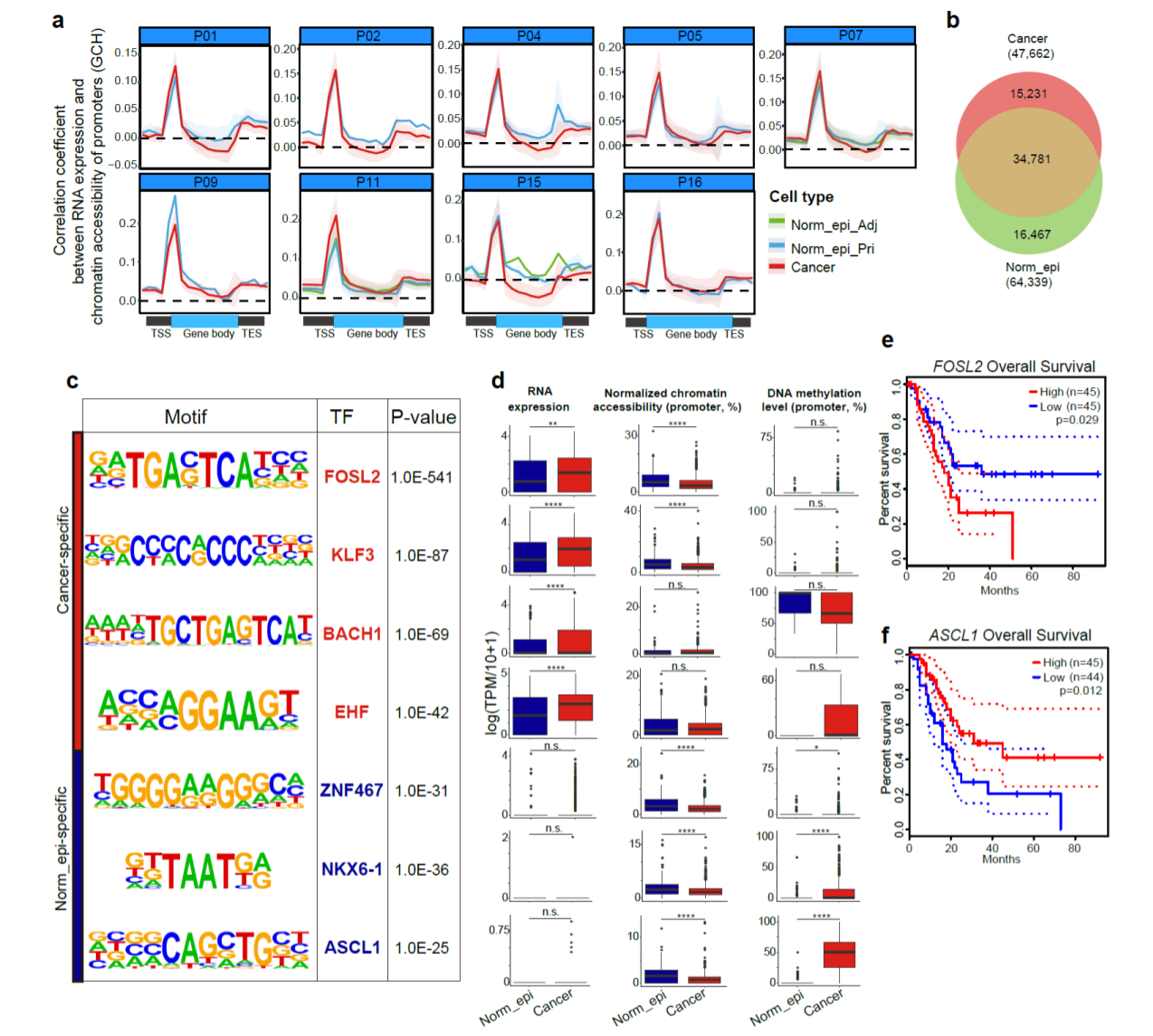
Fig. 5 Open chromatin regions and corresponding TFs in PDAC tumorigenesis.
6. ZNF667 and ZNF667-AS1 as candidate novel markers for prognosis in PDAC.
With integrated promoter methylation, accessibility and RNA expression comparisons, we identified a couple of known and novel prognosis markers for PDAC, ZNF667 and ZNF667-AS1(Fig. 6). We further conducted survival analysis using tissue microarrays with ZNF667 antibody on 98 PDAC patients. Both overall and progression free survival time were significantly longer in patients with higher expression levels of ZNF667. We also separately overexpressed these two genes in two cancer cell lines, PANC-1 and SW1990 and found that both suppressed tumorigenesis of PDAC via suppressing cell proliferation in cancer cells but not via promoting apoptosis.
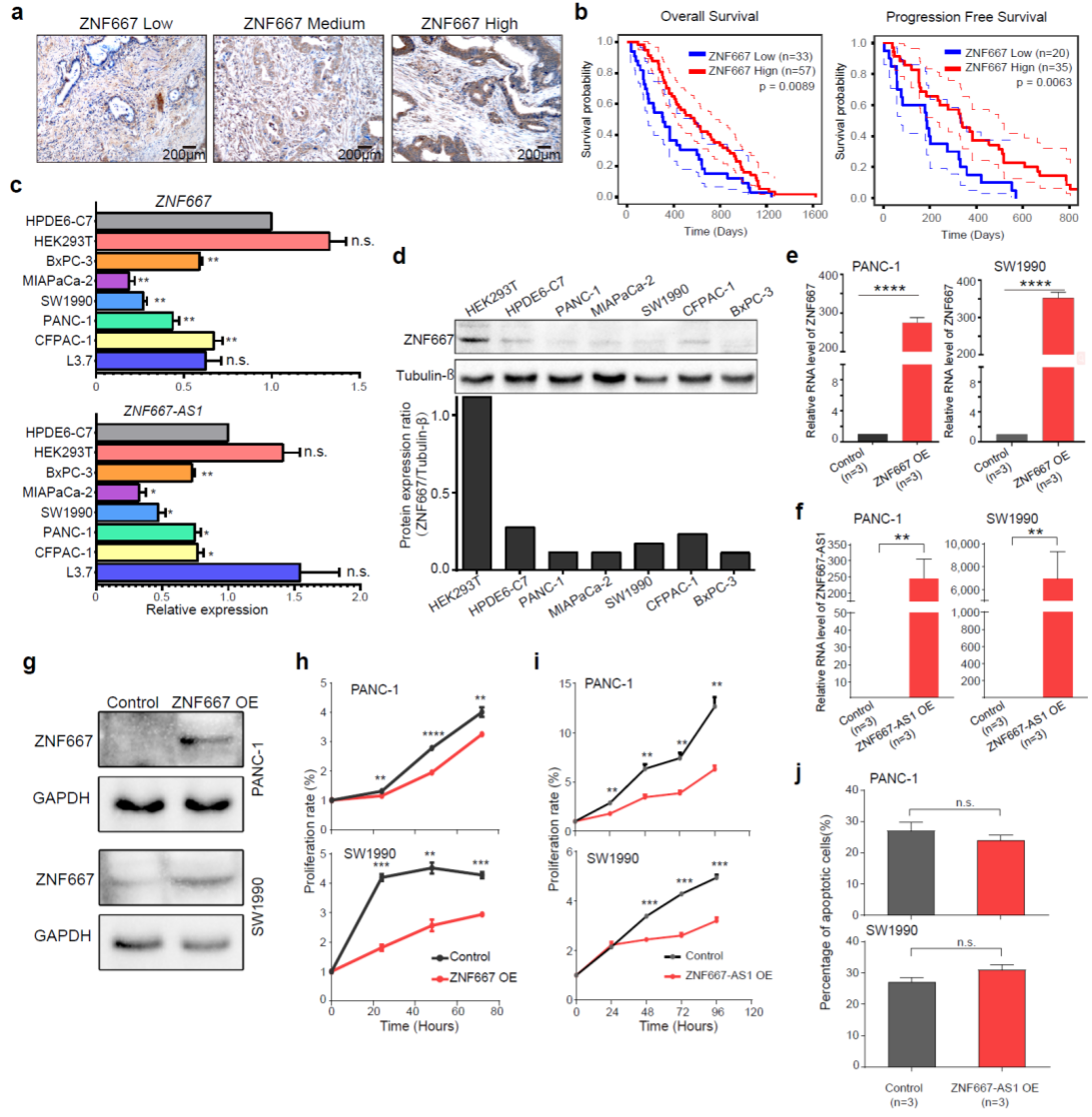
Fig. 6 ZNF667 and ZNF667-AS1 as candidate novel markers for prognosis in PDAC.
In summary, our work offers novel insights into the molecular characteristics of PDAC tumorigenesis, and identifies candidate marker genes for the prognosis of PDAC through integrated single-cell multiomics analyses of PDAC patients.
Dr. Xiaoying Fan from Guangzhou Laboratory, Ph.D. candidate Ping Lu from School of Life Sciences of Peking University, Dr. Hongwei Wang from Tianjin University Cancer Institute, Dr. Shuhui Bian from Nanjing Medical University and Dr. Xinglong Wu from Hebei Agricultural University are the co-first authors of this paper. Professor Jihui Hao from Tianjin University Cancer Institute and Professor Fuchou Tang from Beijing Advanced Innovation Center for Genomics, Biomedical Pioneering Innovation Center, School of Life Sciences of Peking University are corresponding authors. This research project was supported by the National Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics.