Genome Biology | A novel single-cell genome sequencing from Tang Lab, Peking University

On June 30th, 2021, Tang Lab from Biomedical Pioneering Innovation Center (BIOPIC) and Beijing Advanced Innovation Center for Genomics (ICG) published an article named SMOOTH-seq: single-cell genome sequencing of human cells on a third generation sequencing platform on Genome Biology, which developed a novel single-cell genome sequencing technique on third-generation sequencing platform.
Single-cell whole-genome sequencing (scWGS) is a powerful tool to reveal cellular heterogeneity in biological samples and identify genomic changes such as copy number variations (CNVs) and point mutations. Thus, the technology makes it possible to explore the cell lineages, especially the evolution of cells during tumorigenesis, and precisely digs out the information on cellular heterogeneity which is lost in bulk sequencing. Several single-cell genome amplification techniques have been reported previously, such as DOP-PCR, multiple displacement amplification (MDA), multiple annealing and looping-based amplification cycles (MALBAC), and Linear Amplification via Transposon Insertion (LIANTI). However, current scWGS methods are all based on next-generation sequencing platforms generating highly accurate but relatively short reads (several hundred base pairs), which are well-suited for calling copy number variations (CNVs), small indels, and single-nucleotide variations (SNVs), but not optimal for structural variations (SVs).
Several key conclusions emerge from this work:
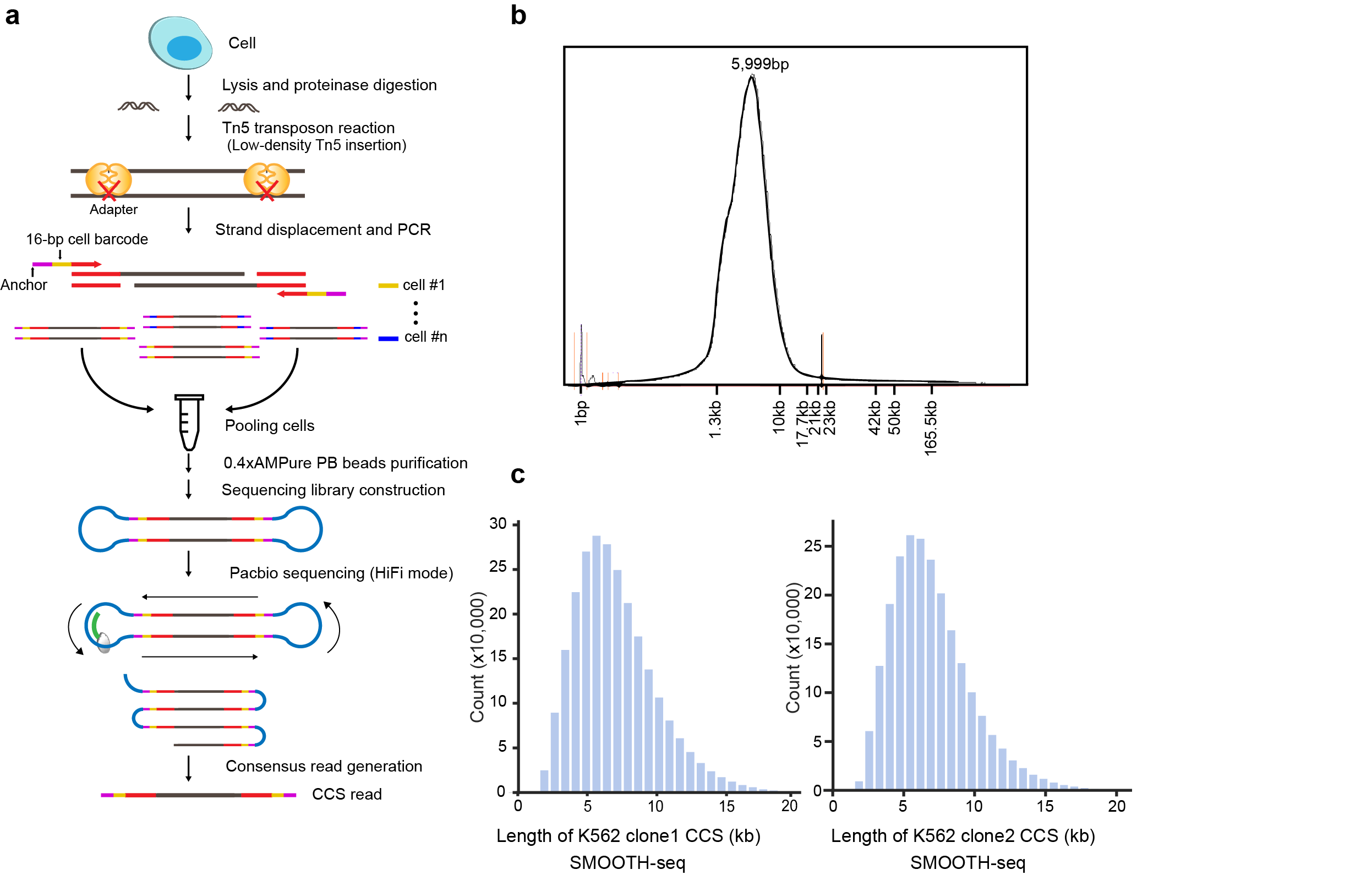
This work developed a novel third-generation sequencing platform-based single-cell whole-genome sequencing (scWGS) method named SMOOTH-seq (single-molecule real-time sequencing of long fragments amplified through transposon insertion). The length of CCS (circular continuous consensus)was over 1kb and could reach as long as 43,693 bp, with the majority of the reads of about 6kb.(Fig.1)
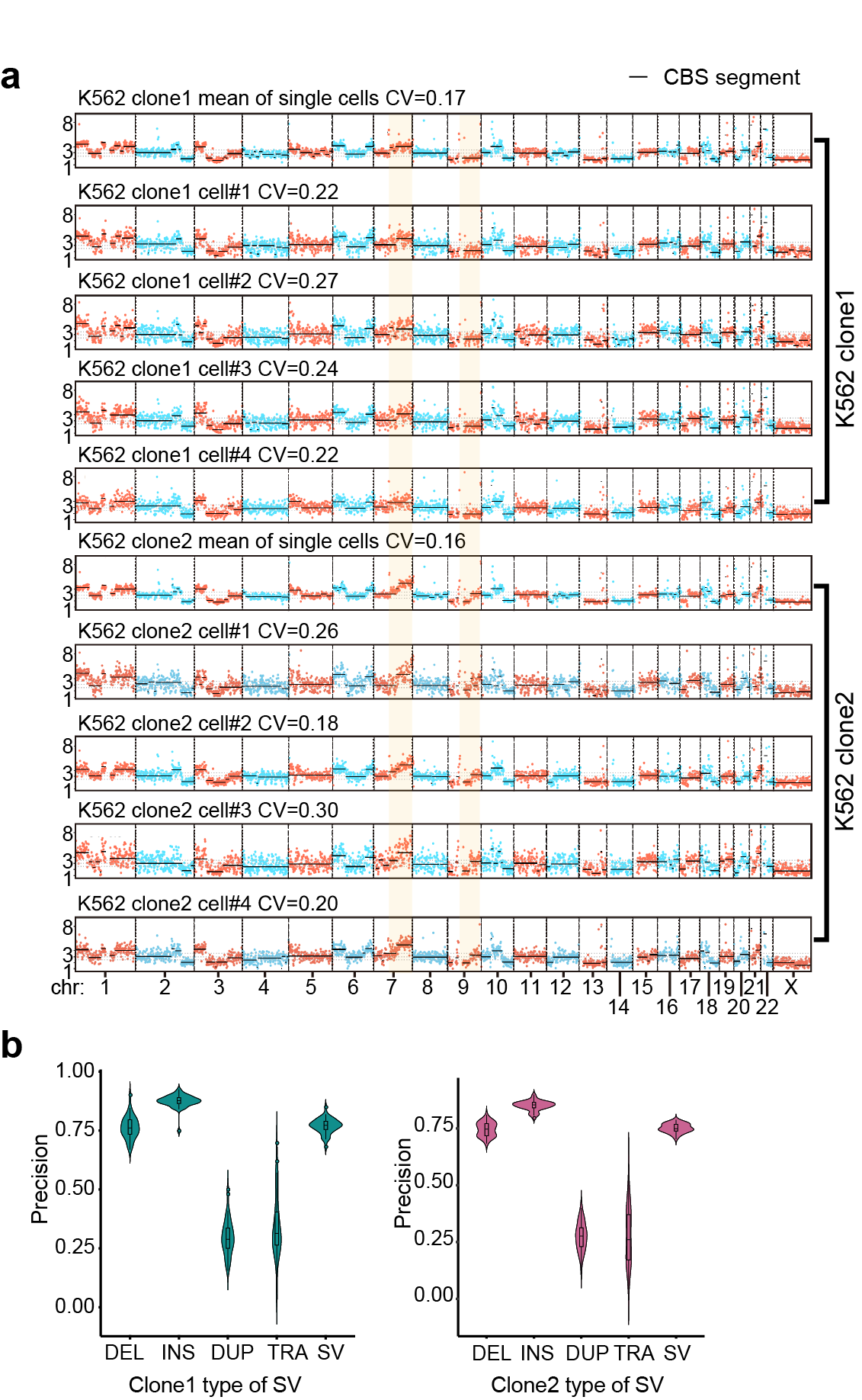
2. Even with relative low genome coverage (0.4X) and sequencing depth, SMOOTH-seq could accurately and stably detect different CNVs between two K562 subclones at 1Mb resolution. And SMOOTH-seq detected SVs in single cells and identified 4,790 deletions and 5,589 insertions, over 87%(91%)of which were less than 1kb long. SMOOTH-seq directly caught the whole inserted fragments as long as 7,780 bp. Moreover, this work observed 521 translocation events in K562 including two classical fusion genes: BCR-ABL and NUP214-XKR3. We further characterized 485 duplication events in K562 as enriched in genomic regions near the telomeres. (Fig.2)

3. SMOOTH-seq identified 125 extrachromosomal circular DNA (ecDNAs) in K562 cells, which ranged from 5 kb to 1 Mb. The long read length of SMOOTH-seq made it possible to capture the full-length ecDNAs less than 10 kb in a single sequencing read. When only one copy of Tn5 transposase binding on an ecDNA molecule, the whole circular DNA molecule could be amplified into one linear fragment that cover its full-length sequence. Sanger sequencing validated 90% of selected ecDNAs. About one third (29.6%) of the ecDNAs contained genes and they enriched with GO terms associated with cellular identity and cell cycle, indicating functional regulation of ecDNAs (Fig.3) .
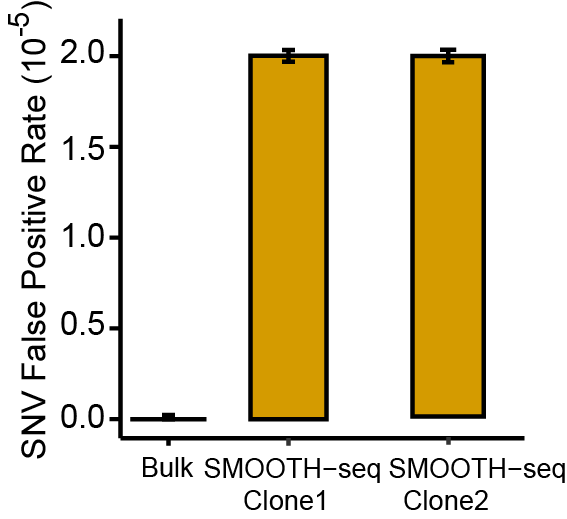
4. The false-positive rate for SNV calling in an individual cell was 2.0 × 10-5 using SMOOTH-seq and the spectra of false positives showed little preference comparing to those of bulk. (Fig.4)
5. SMOOTH-seq accurately detected structural variations events in colorectal cancer tumor samples. By identifying the SVs supported by at least 2 colorectal cancer cells (CRCs), this work identified 4089 insertion events, 3852 deletion events, 341 translocation events, and 312 duplications. Extracting the CRC-specific SV mutations by removing those overlapped with the SVs identified in K562 cells. 3570 SV events (1376 insertions, 1661 deletions, 230 translocations, and 303 duplications) were retained as CRC-specific. After further checking these SVs using PCR in a couple of gDNA samples, including the corresponding tumor tissue, tumor adjacent normal tissue, an B cell line GM12878, and the peripheral blood mononuclear cells from another person, this work found the PCR results were the same in all the gDNA samples, different from the suspected size in the current genome reference, but including the size of each SVs. The results were interpreted as the current human genome annotation being still imperfect and the third-generation sequencing would be helpful in reconstructing a more complete genome reference.
In summary, SMOOTH-seq applies the long-read third-generation sequencing technology to the single-cell genome sequencing, which can detect structural variations, extrachromosomal circular DNA, etc. The high-precision detection of this molecular event greatly increases the scope of application of single-cell genome sequencing technology and has broad application prospects. This research ushered in the era of single-cell genome single-molecule sequencing. The single-cell genome single-molecule sequencing technology developed by this research will uncover more of the mystery of the "dark matter" in the human genome and bring new development opportunities to human biomedical research.
Dr. Xiaoying Fan from Bioland Laboratory, Dr. Cheng Yang and Ph.D. candidate Wen Li from Academy for Advanced Interdisciplinary Studies of Peking University are the co-first authors of the paper. Professor Fuchou Tang of Beijing Advanced Innovation Center for Genomics, Biomedical Pioneering Innovation Center, School of Life Sciences of Peking University is corresponding author. This research project was supported by the National Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics.
Link: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02406-y