Cancer Cell | Fuchou Tang's research group cooperate with Wei Fu's research group to reveal the prevalent genomic alterations in tumor microenvironment cells of human colorectal cancer
The tumor microenvironment (TME) is composed of various non-epithelial cells and extracellular matrix. The non-epithelial cells mainly include tumor-infiltrating immune cells, fibroblasts, and vascular endothelial cells. Tumor-infiltrating immune cells include T cells, B cells, NK cells and macrophages, etc. The complex interactions between these non-malignant cells and cancer epithelial cells contribute to the progression of tumor and contain many clues for cancer treatment. In recent years, single-cell transcriptome sequencing methods have been widely used in the researches of the TME, but to what extent these microenvironmental cells are transformed by cancer cells, especially whether the TME cells have undergone genetic alterations deserves further investigations.
Recently, several studies have confirmed that gene mutations are pervasive in the epithelial cells of human normal tissues, and the accumulation of mutations in the epithelial cells of "normal" tissues is considered to be a potential precancerous mechanism [1]. However, previous researches on large-scale somatic copy number alterations (SCNAs) using bulk sequencing can only detect clonal SCNAs with high frequency (>5%) in only a small proportion of individuals due to technical limitations, thus ignored the potential low-frequency, non-clonal SCNAs and greatly underestimated the prevalence of SCNAs in human "normal" tissues [2, 3].

On October 22, 2020, Fuchou Tang’s research group from the Biomedical Pioneering Innovation Center of Peking University (BIOPIC), Beijing Advanced Innovation Center for Genomics (ICG), the School of Life Sciences in Peking University and Wei Fu’s research group from Department of General Surgery, Peking University Third Hospital published a research paper entitled "Single-cell Multiomics Sequencing Reveals Prevalent Genomic Alterations in Tumor Stromal Cells of Human Colorectal Cancer" on Cancer Cell.
In this study, primary tumors, lymph nodes, adjacent normal tissues and peripheral blood samples were obtained from 21 microsatellite-stable colorectal cancer (CRC) patients. Also, peripheral blood samples were obtained from 6 cancer-free individuals. High-precision parallel single-cell genome and transcriptome sequencing was conducted on multiple types of non-epithelial cells. Firstly, a single-cell multiomics sequencing method has been developed and optimized. The cytoplasm and nucleus of a single cell were separated from each other, enabling simultaneous transcriptome and genome sequencing of the same single cell. Based on the method, the frequencies and patterns of SCNAs in immune cells, fibroblasts and vascular endothelial cells in both the TME and normal tissues have been systematically studied, revealing that fibroblasts with SCNAs from the TME underwent specific clonal expansion process. Also, five fibroblast-specific markers in the TME were discovered through single-cell transcriptome data analysis, which were associated with poorer prognosis of CRC (Figures 1 and 2).
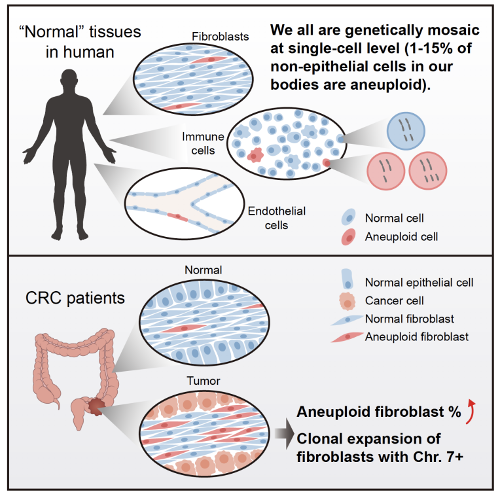
Figure 1. Summary of the main findings in the study
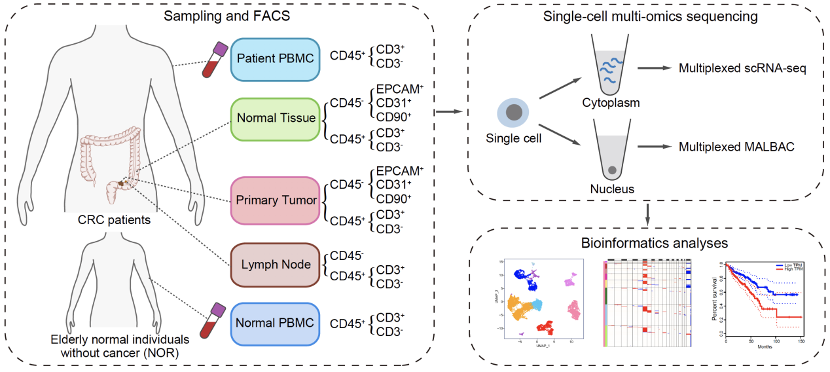
Figure 2. Study design for sampling, sequencing and bioinformatics analysis
The study had 3 major findings:
(1) Somatic copy number alterations were pervasive in various immune cells, fibroblasts and vascular endothelial cells in normal human tissues.
In this study, single-cell multiomics sequencing method was conduct on a total of 15,312 single cells from 21 CRC patients and 6 cancer-free normal individuals. These cells were classified into 8 major cell types according to known markers, including epithelial cells, fibroblasts, endothelial cells, T cells, NK cells, B cells, mast cells and other myeloid cells. Surprisingly, even in non-cancerous normal tissues, 1%-15% of all the cell types were found to have SCNAs (>10 Mb). It is very possible that a few percent of all the major cell types in human body have undergone genetic alterations including SCNAs. Genetic abnormalities are not limited to epithelial cells in human body, but are pervasive in all major cell types. These cells with permanent and irreversible genetic abnormalities may be involved in the occurrence and development of many human diseases.
Based on the single-cell genome data, the researchers found that genetically abnormal immune cells with SCNAs not only exist in the tumor-infiltrating immune cells (1.3%-11.2%), but also in the adjacent “normal” tissue-infiltrating immune cells (0.9%-9.7%), the immune cells from peripheral blood of CRC patients (1.1%-11.5%) and the immune cells from peripheral blood of cancer-free individuals (2.9%-10.2%). A total of 538 (6.0%) cells with SCNAs were identified from all the 8,982 immune cells that passed strict genomic data quality control. SCNAs was pervasive in the immune cells of every colorectal cancer patient (3.0%-11.2%) and every cancer-free individual (2.9%-10.2%). By exploring the chromosomal features of these abnormal immune cells, the researchers found that in female individuals, the loss of the entire X chromosome occurred most frequently (one of the two X chromosomes is missing in a single cell, changing from XX to XO), and in male individuals, the loss of the entire Y chromosome occurred most frequently (changing from XY to XO), while the copy number variation of autosomes seemed to be randomly distributed (Figure 3). Vascular endothelial cells with SCNAs were also common in adjacent normal tissues (0.0%-1.4%), blood vessels in the colon mesentery (3.7%-14.9%) and primary tumors (3.9%-12.8%).
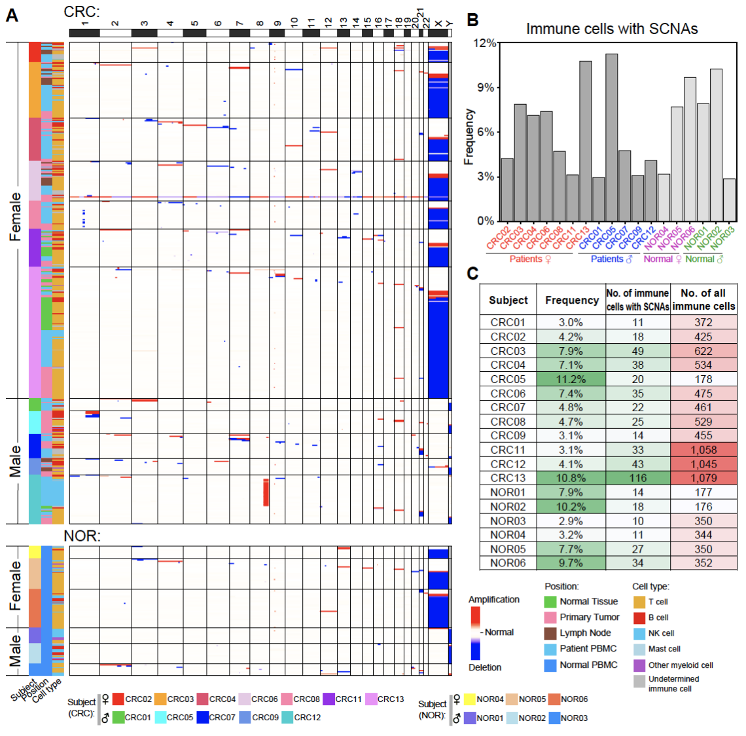
Figure 3. Immune cells with SCNAs are pervasive in all the individuals involved
(2) The frequencies of fibroblasts with SCNAs in primary tumors were much higher than those in adjacent normal tissues and the genetically abnormal fibroblasts have undergone clonal expansion in tumor tissues.
A total of 37 fibroblasts with SCNAs from adjacent normal tissues (out of 664 fibroblasts from 7 CRC patients) and 333 fibroblasts with SCNAs from primary tumors (out of 1,183 fibroblasts from 9 CRC patients) were identified. The frequency of fibroblasts with SCNAs from primary tumors in each patient varied from 11.1% to 47.7%, which was about 4 times higher than that from adjacent normal tissues (1.1%-11.5%) (Figure 4).
In addition, SCNAs of fibroblasts in adjacent normal tissues showed a relatively random distribution, while fibroblasts with copy number gain of chromosome 7 were significantly enriched in tumor tissues. Among 88 fibroblasts from tumor tissues with copy number variation of chromosome 7, 85 cells had an increase in the copy number of the entire chromosome 7, and the average expression level of genes located on chromosome 7 in these 85 cells was significantly higher (26% increase) than fibroblasts without SCNAs. The specific enrichment of the increased copy number of Chromosome 7 suggests the clonal expansion of fibroblasts with SCNAs in the TME (Figure 5). It shows that the TME cells are not simply "induced" by cancer epithelial cells and passively participate in tumorigenesis, but have already undergone irreversible and permanent genetic alterations, and may actively promote tumorigenesis. Genetic variations do not only exist in tumor epithelial cells, but also widely exist in non-epithelial TME cells.
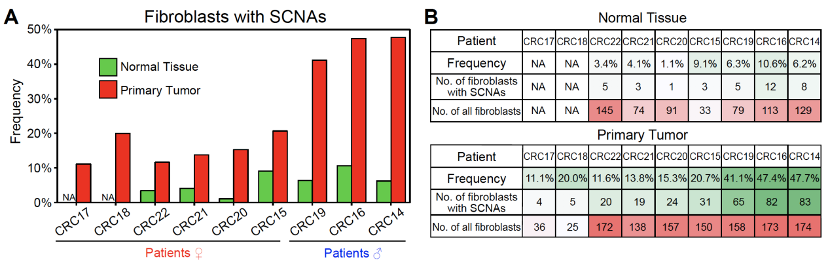
Figure 4. The frequencies of fibroblasts with SCNAs in primary tumors are much higher than those in adjacent “normal” tissues
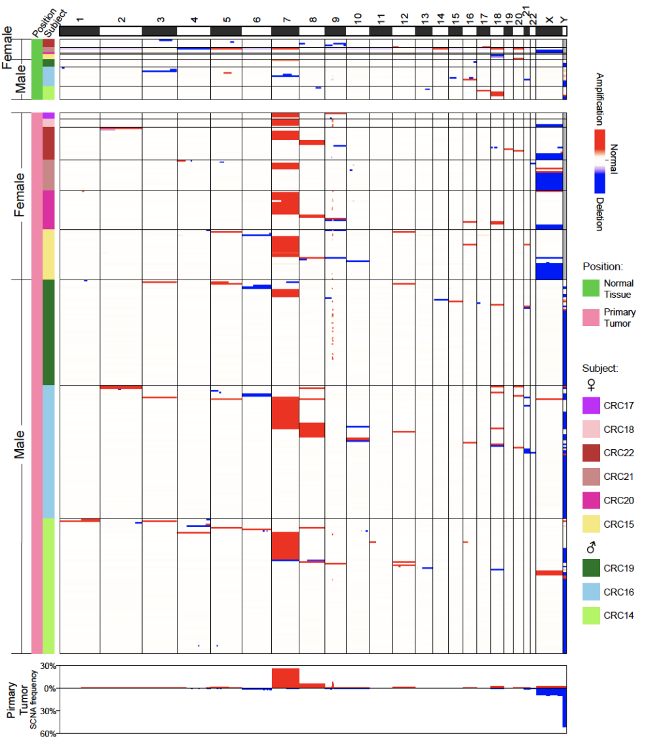
Figure 5. Fibroblasts with copy number gain of chromosome 7 are significantly enriched in tumor tissues
(3) Five fibroblast-specific markers in the TME were identified through single-cell transcriptome data analysis, whose higher expressions were associated with poorer prognosis of CRC.
A set of significantly differentially expressed genes (DEGs) between fibroblasts from primary tumors and fibroblasts from adjacent normal tissues were identified, including 59 up-regulated genes and 17 down-regulated genes in fibroblasts from primary tumors. Among them, BGN, RCN3, TAGLN, MYL9 and TPM2 were significantly up-regulated in fibroblasts from tumor tissues, and the high expression of these genes was verified to clearly related to poorer prognosis in TCGA database of CRC. Immunohistochemical staining were performed to further verify that these 5 genes were specifically and highly expressed in fibroblasts from tumor tissues (Figure 6). This discovery expands the types of tumor marker genes. The original marker genes are mainly genes specifically expressed in cancer epithelial cells. Genes that specifically expressed in the TME cells can also be tumor marker genes and are more likely to have universal applicability across different cancer types.
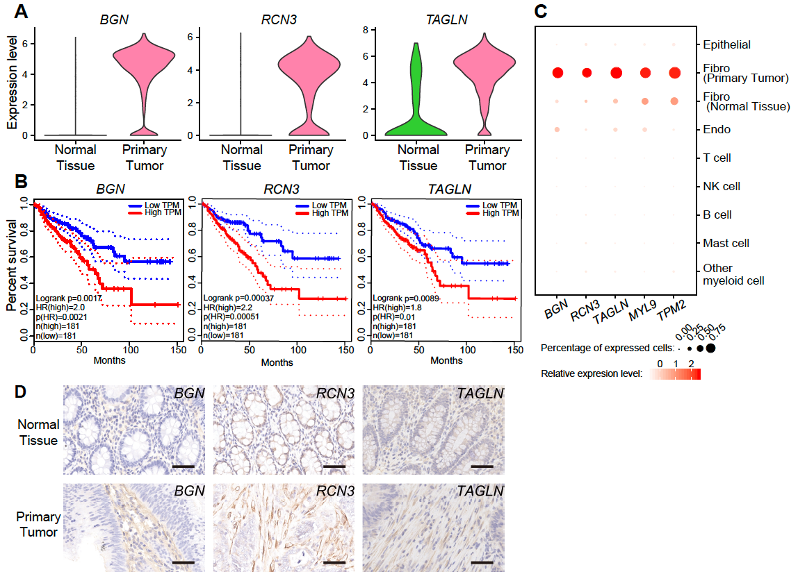
Figure 6. Upregulated genes in fibroblasts from primary tumors indicate poorer prognosis of CRC
In summary, this study used high-precision single-cell multi-omics sequencing technology to thoroughly and systematically analyze the somatic copy number alterations of various non-epithelial cells obtained from 27 human individuals, revealing that SCNAs were pervasive in fibroblasts, immune cells and endothelial cells. The proportion of fibroblasts with SCNAs in the TME of CRC greatly increased, and clonal expansion of these genetically abnormal fibroblasts were found in tumors. This research breaks through people’s understanding of genetic alterations in human "normal" tissues, and provides convincing evidence that genetic alterations, especially SCNAs are pervasive in the population and in the major cell types of the human body. The results suggest that copy number alterations may play an important role in human diseases and aging, and provide new clues for tumor microenvironment and tumor treatment.
Ph.D. candidate Yuan Zhou, Shuhui Bian Ph.D. and Ph.D. candidate Yueli Cui from Biomedical Pioneering Innovation Center of Peking University (BIOPIC) and Xin Zhou Ph.D. from Department of General Surgery, Peking University Third Hospital are the co-first authors of the paper. Professor Fuchou Tang and Professor Wei Fu are the co-corresponding authors of the paper. The work is supported by grants from the National Natural Science Foundation of China and Beijing Advanced Innovation Center for Genomics (ICG).
References:
1. Yizhak, K. et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, aaw0726, (2019).
2. Jacobs, K. B. et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 44, 651-658, (2012).
3. Laurie, C. C. et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 44, 642-650, (2012).